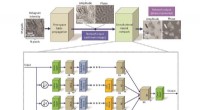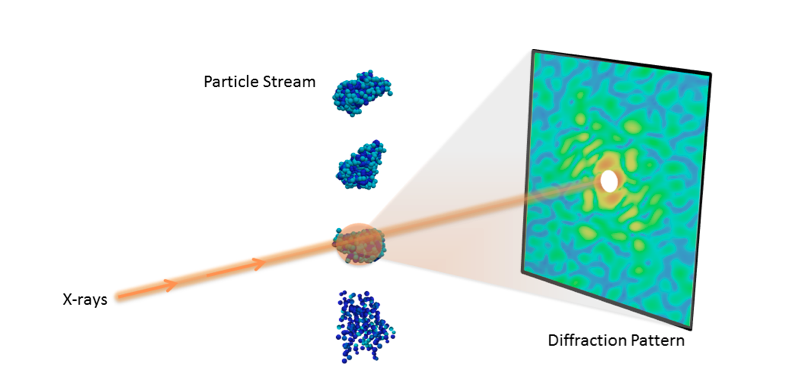
Montage expérimental pour une expérience de diffraction à une seule particule. Crédit :Peter Zwart, Laboratoire de Berkeley
Comprendre la structure moléculaire 3D d'importants nano-objets tels que les protéines et les virus est crucial en biologie et en médecine. Avec les progrès récents de la technologie des rayons X, les scientifiques peuvent désormais collecter des images de diffraction de particules individuelles, permettant finalement aux chercheurs de visualiser des molécules à température ambiante.
Cependant, déterminer la structure 3D à partir de ces expériences de diffraction de particules uniques est un obstacle important. Par exemple, les taux d'acquisition de données actuels sont très limités, résultant généralement en moins de 10 instantanés utiles par minute, limiter le nombre de fonctionnalités pouvant être résolues. En outre, les images sont souvent fortement corrompues par du bruit et d'autres artefacts expérimentaux, rendant difficile l'interprétation correcte des données.
Pour relever ces défis, une équipe de chercheurs du Lawrence Berkeley National Laboratory (Berkeley Lab) a développé un nouveau cadre algorithmique appelé phasage itératif à plusieurs niveaux (M-TIP) qui utilise des techniques mathématiques avancées pour déterminer la structure moléculaire 3D à partir d'ensembles très clairsemés de bruit, données sur une seule particule. Cette approche permet essentiellement aux chercheurs d'extraire plus d'informations à partir d'expériences avec des données limitées. Les mathématiciens appliqués Jeffrey Donatelli et James Sethian, et le bioscientifique physique Peter Zwart ont introduit ce cadre en développant un algorithme qu'ils ont développé à l'origine pour résoudre la reconstruction à partir d'une expérience de diffusion des rayons X connexe, appelée diffusion des rayons X par fluctuation. Un article décrivant le cadre M-TIP a été publié le 26 juin dans le Actes de l'Académie nationale des sciences .
« Cette approche a le potentiel de révolutionner le domaine, " dit Zwart. " Étant donné qu'il est difficile d'obtenir beaucoup de bonnes données, les approches qui réduisent la quantité de données nécessaires pour imager avec succès des nano-objets 3D sont susceptibles de recevoir un accueil chaleureux. »
Donatelli, Sethian et Zwart font tous partie de CAMERA (The Center for Advanced Mathematics for Energy Research Applications), dont la mission est de créer les mathématiques de pointe nécessaires pour traiter les données de plusieurs des installations scientifiques les plus avancées du DOE. CAMERA est financé conjointement par les programmes Advanced Scientific Computing Research et Basic Energy Sciences du Bureau des sciences du DOE.
Diffraction de particule unique
L'avènement récent des lasers à électrons libres à rayons X (XFEL) a permis plusieurs nouvelles techniques expérimentales pour étudier les biomolécules qui étaient infaisables avec les sources lumineuses traditionnelles. Une de ces techniques est la diffraction à une seule particule, qui recueille un grand nombre d'instantanés de diffraction des rayons X avec une seule particule dans le faisceau. En exploitant la puissance extrême des XFEL, les chercheurs peuvent collecter des signaux mesurables même à partir des plus petites particules.
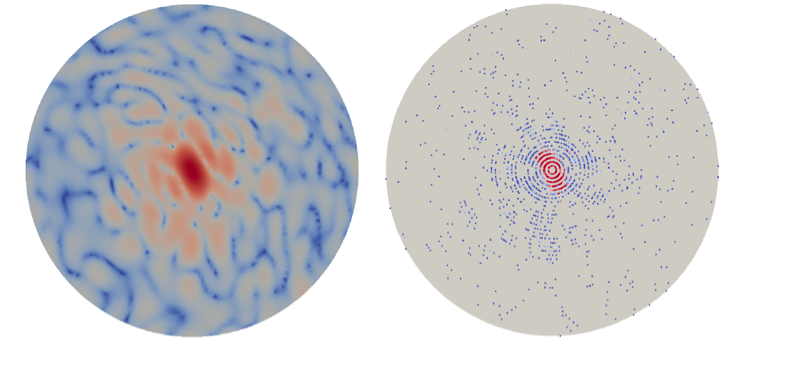
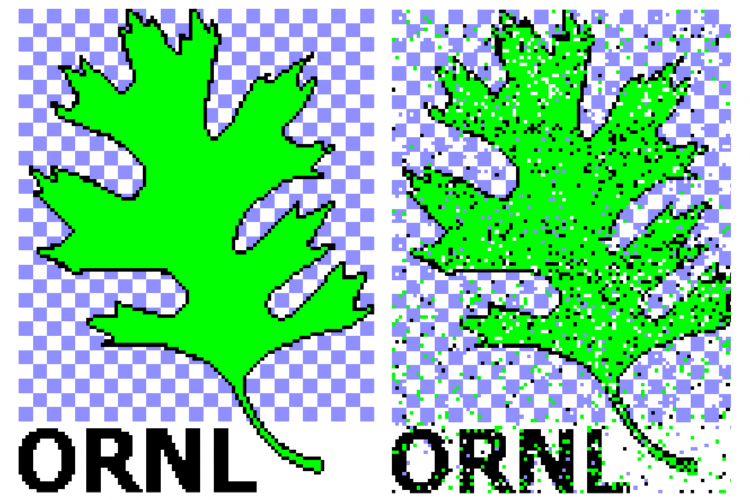
Un exemple d'une image de diffraction propre à une seule particule (à gauche) et la même image de diffraction après contamination par le bruit (à droite). Crédit :Peter Zwart, Laboratoire de Berkeley
Un grand avantage offert par cette technique de diffraction à particule unique est la possibilité d'étudier comment différentes copies d'une molécule varient ou changent de forme. Puisque chaque image provient d'une seule particule, ces variations peuvent être capturées dans l'expérience, contrairement aux méthodes d'imagerie traditionnelles comme la cristallographie ou la diffusion des rayons X aux petits angles, où les chercheurs ne peuvent mesurer qu'une moyenne sur tous les différents états de l'échantillon moléculaire.
Cependant, déterminer la structure 3D à partir de données de diffraction de particules uniques est un défi. Pour commencer, lorsque chaque particule est imagée, son orientation est inconnue et doit être récupérée afin de combiner correctement les données dans un volume de diffraction 3D. Ce problème est aggravé si la molécule peut prendre différentes formes, ce qui nécessite une classification supplémentaire des images. Par ailleurs, les informations de phase ne sont pas enregistrées dans les images de diffraction et doivent être récupérées afin de terminer la reconstruction. Finalement, même avec des XFEL puissants, le nombre de photons diffusés est très faible, résultant en des images extrêmement bruitées, qui peuvent être davantage contaminés par des problèmes systématiques de fond et de lecture du détecteur.
Les approches précédentes sont basées sur la résolution du problème de reconstruction en étapes séparées, où chaque problème individuel est traité séparément. Malheureusement, un inconvénient de ces approches en série est qu'elles n'exploitent pas facilement les caractéristiques connues antérieures sur l'apparence de la molécule. En outre, toute erreur commise dans une étape est propagée à la suivante, résultant en une nouvelle augmentation de l'erreur. Cette « erreur boule de neige » dégrade finalement la qualité de la reconstruction obtenue à l'étape finale.
Le meilleur des deux mondes
Au lieu de résoudre les problèmes de calcul en étapes séparées, l'algorithme M-TIP de l'équipe résout toutes les parties du problème simultanément. Cette approche exploite les informations préalables sur la structure pour réduire considérablement les degrés de liberté du problème à toutes les étapes, et par conséquent réduire les informations requises pour réaliser une reconstruction 3D.
"Les techniques d'optimisation de boîte noire standard peuvent incorporer des connaissances préalables dans la reconstruction mais rejeter toute la structure du problème, alors que le résoudre en sous-étapes en série complètement séparées exploite la structure du problème mais jette presque toutes les informations antérieures sur ce à quoi pourrait ressembler la solution, " a déclaré Donatelli. " M-TIP tire parti du meilleur des deux mondes en exploitant la structure du problème pour diviser le calcul en plusieurs morceaux gérables, puis en affinant itérativement tous ces morceaux pour arriver à une solution qui soit cohérente avec les deux données et des contraintes structurelles.
En utilisant cette technique, l'équipe a pu déterminer la structure 3D à partir d'un nombre extrêmement faible d'images à partir de données simulées, aussi peu que 6 à 24 images pour des données sans bruit et 192 images pour des données hautement contaminées.
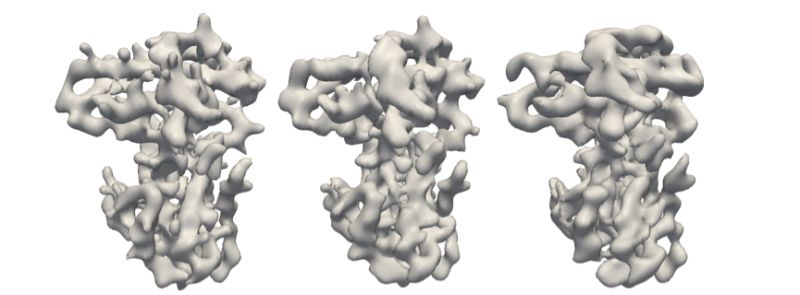
Protéine de rétinoblastome originale (à gauche) et reconstructions utilisant l'algorithme M-TIP avec 24 images nettes (au milieu) et 192 images bruitées (à droite), comme le montre la figure 2. Crédit :Peter Zwart, Laboratoire de Berkeley
Innover
Ce travail fait partie d'une nouvelle initiative de collaboration entre le SLAC National Accelerator Laboratory, CAMÉRA, le Centre national de calcul scientifique de la recherche énergétique (NERSC) et le Laboratoire national de Los Alamos dans le cadre du projet Exascale Computing (ECP) du DOE. L'objectif du projet est de développer les outils de calcul nécessaires pour effectuer une analyse de données en temps réel à partir d'expériences menées à la source de lumière cohérente Linac (LCLS) du SLAC. Avec des mises à niveau de la ligne de lumière, LCLS-II prévoit de générer plusieurs téraoctets de données par seconde, lequel, par exemple, permettra aux scientifiques de développer considérablement les expériences actuelles sur une seule particule. L'analyse de toutes ces données en temps réel nécessitera de nouveaux algorithmes et de grosses machines de calcul. L'algorithme M-TIP fera partie de ce processus.
« Ce sont quelques-uns des problèmes les plus difficiles de la science des données informatiques, " dit Sethian. " Pour les attaquer, nous devons exploiter une gamme de technologies, y compris les architectures informatiques exascale émergentes, des réseaux haut débit sophistiqués, et les algorithmes mathématiques les plus avancés disponibles. Réunir les scientifiques de CAMERA avec des projets d'application exascale a ouvert la porte à la construction d'outils pour aborder certains problèmes urgents de la biologie et des sciences des matériaux."
Les chercheurs notent que ce ne sont que les premières étapes. Pour que la méthode soit prête à être déployée, d'autres obstacles doivent être surmontés.
"La science expérimentale est désordonnée, " dit Zwart. " Il y a des effets expérimentaux supplémentaires qui doivent être pris en considération pour que nous obtenions les meilleurs résultats possibles. "
"Heureusement, M-TIP est une technique très modulaire, " ajoute Donatelli, "donc, il est bien adapté à la modélisation de bon nombre de ces effets supplémentaires sans avoir besoin de changer le cadre algorithmique de base. »
L'équipe travaille actuellement à l'étude de ces effets dans le cadre de la Single Particle Initiative, un grand, collaboration multi-institutionnelle dédiée à la résolution des problèmes théoriques et pratiques de l'imagerie moléculaire unique basée sur le X-FEL, aboutissant à terme à fournir à la communauté scientifique les outils nécessaires pour innover en biologie, médecine et sciences de l'énergie.