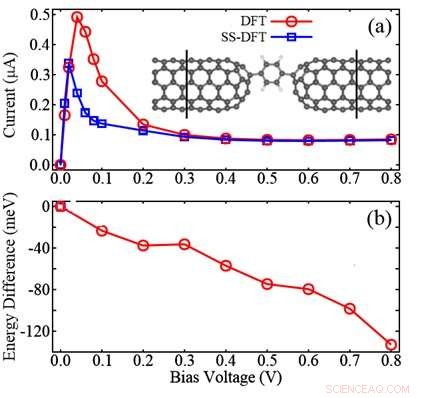
La figure montre la comparaison entre les méthodes SS-DFT et les méthodes DFT largement utilisées pour un dispositif moléculaire composé de deux électrodes de nanotubes de carbone (CNT) et d'une molécule de benzène entre :(a) courbes I-V calculées ; (b) la différence d'énergie calculée en soustrayant l'énergie DFT de celle SS-DFT. La figure montre que SS-DFT prédit l'état de transport énergétiquement plus stable avec des courants électriques inférieurs par rapport à la méthode basée sur DFT. Crédit :Zhang Chun
Les informaticiens de NUS ont développé une nouvelle version de la théorie fonctionnelle de la densité (DFT) pour étudier les dispositifs à l'échelle nanométrique.
Les appareils électroniques sont de plus en plus petits et intègrent de plus en plus de fonctionnalités. Ceci est rendu possible en réduisant la taille des composants électroniques. Lorsque leur taille diminue, les propriétés de ces dispositifs moléculaires deviennent beaucoup plus sensibles à leur environnement extérieur. Des méthodes de calcul sont nécessaires pour simuler et prédire les propriétés de ces petits dispositifs. L'un d'eux est la théorie de la fonctionnelle de la densité. Ces méthodes sont développées à partir de principes premiers, comprenant des connaissances de base et fondamentales que nous connaissons déjà. Les informaticiens de NUS ont affiné la théorie pour prendre en compte les effets de non-équilibre présents lors du fonctionnement des appareils (par exemple, lorsqu'une batterie est connectée à un système quantique). Cela conduit à une prédiction plus précise et plus précise.
Le professeur ZHANG Chun et son doctorat étudiant, LIU Shuanglong avec un chercheur, Dr Argo NURBAWONO, du Département de physique, NUS a développé une version plus générale de la théorie de la fonctionnelle de la densité (DFT) populaire et largement utilisée qui peut être appliquée à des situations de non-équilibre en régime permanent. Ils ont introduit un degré de liberté supplémentaire, connue sous le nom de densité électronique de non-équilibre, dans la modélisation des premiers principes. Cela prend en compte les effets de non-équilibre induits par le biais lorsqu'un dispositif moléculaire fonctionne sous un biais fini. Cette nouvelle version de la théorie est connue sous le nom de DFT en régime permanent (SS-DFT).
Les chercheurs ont montré que la DFT largement utilisée en principe n'est pas correcte dans un scénario de non-équilibre en régime permanent. Dans une telle situation, deux paramètres différents, la densité électronique totale et la densité d'électrons porteurs de courant, sont nécessaires pour déterminer les propriétés du système de non-équilibre correspondant. La nouvelle théorie a été mise en œuvre dans le progiciel de calcul de l'Initiative espagnole pour les simulations électroniques avec des milliers d'atomes (SIESTA) pour étudier les propriétés électroniques/de transport de divers dispositifs à l'échelle moléculaire.
Le SS-DFT fournit un outil théorique fiable pour la compréhension et la conception future de nouveaux dispositifs à l'échelle moléculaire avec des fonctionnalités améliorées. Le progiciel de calcul basé sur SS-DFT est maintenant utilisé par de nombreux groupes de recherche dans le monde entier. Il est utilisé pour expliquer des phénomènes de transport intrigants observés expérimentalement au niveau moléculaire et pour concevoir de nouveaux types de dispositifs moléculaires.
Les chercheurs prévoient d'étendre l'applicabilité de la théorie en incluant d'autres effets physiques, telles que les interactions électron-phonon et le couplage spin-orbital. Ils ont également l'intention d'améliorer l'efficacité de calcul afin qu'il puisse être utilisé pour modéliser de grands systèmes autour de 1, 000 atomes.