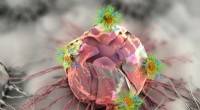
Une illustration montre une protéine d'histocompatibilité majeure (grise) englobant un peptide tiré d'un virus SARS-CoV (rose). Le complexe aide à déclencher l'activation des cellules T qui font partie du système immunitaire. Des chercheurs de l'Université Rice ont découvert un résidu de liaison non-ancre dans le peptide qui pourrait à la fois contribuer à la liaison et à l'activation des lymphocytes T nécessaire pour vaincre le virus. Crédit :Kavraki Lab/Université du riz
La "position 4" ne semblait pas importante jusqu'à ce que les chercheurs se penchent longuement sur un peptide particulier.
Cette partie du peptide extraite d'un virus du SRAS-CoV s'est avérée avoir une influence inattendue mais significative sur la façon dont elle se lie de manière stable à un récepteur central pour la capacité du système immunitaire à attaquer les cellules malades.
Dans une étude publiée par le Actes de l'Académie nationale des sciences , des chercheurs de la Brown School of Engineering de l'Université Rice et du MD Anderson Cancer Center de l'Université du Texas ont révélé des modèles à résolution atomique qui détaillent non seulement la liaison, mais aussi, pour la première fois, les mécanismes de déliaison qui sous-tendent un élément clé du système immunitaire.
Ils disent qu'une meilleure compréhension de l'ensemble du mécanisme pourrait conduire à des progrès en immunothérapie qui renforcent la capacité du corps à lutter contre la maladie.
L'informaticienne de Rice Lydia Kavraki, l'ancien élève Jayvee Abella et le chercheur postdoctoral Dinler Antunes, dirigé l'étude.
"Trouver de bonnes cibles pour déclencher une réponse immunitaire protectrice est très difficile, en particulier dans la recherche contre le cancer, " a déclaré Antunes. " Le fait que ce peptide particulier ait été prédit ne pas se lier aux HLA (antigènes leucocytaires humains) par des méthodes basées sur la séquence met en évidence un angle mort dans notre capacité de prédiction actuelle.
"En intégrant l'analyse structurelle, nous pouvons détecter la contribution de ces interactions secondaires à la fixation et à la stabilité des peptides, nous permettant, espérons-le, de trouver de meilleures cibles pour le développement de vaccins antiviraux et l'immunothérapie du cancer à base de lymphocytes T, " il a dit.
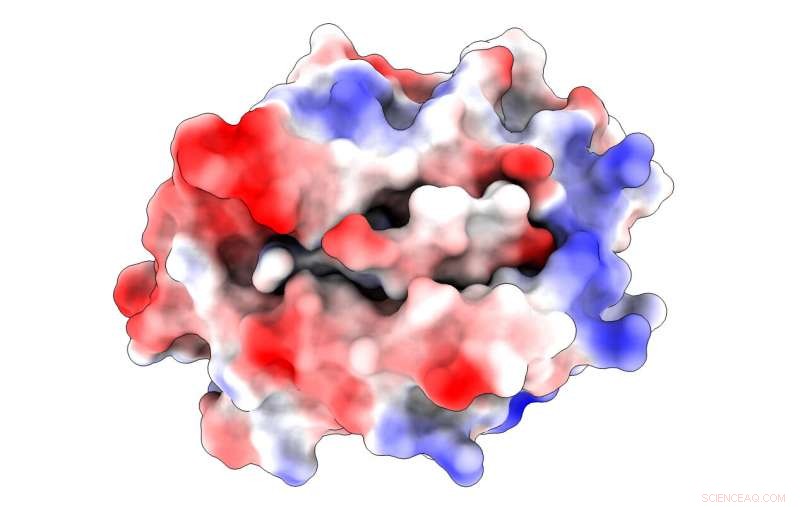
Une illustration montre les charges électrostatiques (le bleu est positif, rouge négatif) dans une protéine majeure d'histocompatibilité liée à un peptide issu d'un virus SARS-CoV. Le complexe aide à déclencher l'infection des cellules T qui font partie du système immunitaire. Des chercheurs de l'Université Rice ont découvert un résidu de liaison non-ancre dans le peptide qui pourrait à la fois contribuer à la liaison et à l'activation des lymphocytes T nécessaire pour vaincre le virus. Crédit :Kavraki Lab/Université du riz
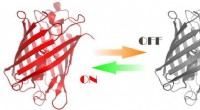
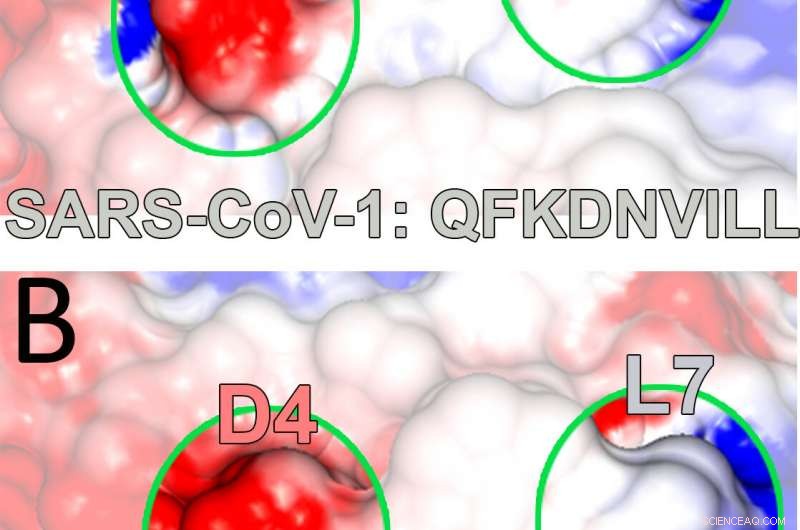
Les chercheurs ont utilisé leurs simulations pour éclairer les détails de la façon dont le peptide intracellulaire du SRAS, QFKDNVILL, se lie à une protéine réceptrice du CMH connue sous le nom de HLA-A*24:02, principalement au niveau des ancres dominantes aux deux extrémités du peptide (aux positions 2 et 9) et les présente pour inspection aux cellules T du système immunitaire.
La liaison stable d'un peptide et du CMH est une condition préalable à l'activation des cellules T, qui recherchent des peptides qui ne se trouvent pas normalement dans les cellules saines. Si le peptide et la protéine ne se lient pas, la cellule T n'est pas incitée à attaquer.
"Cela était connu des études précédentes sur les états liés et non liés de nombreux complexes de ce type, " a déclaré Kavraki. " Ce qu'ils n'ont pas capturé, ce sont les états intermédiaires et les transitions qui mènent d'un état à un autre, surtout le déliement.
"Je pense que c'est la seule analyse qui montre la déliaison des peptides du CMH avec une résolution atomique, " a déclaré Kavraki. " D'autres peptides ont des caractéristiques similaires et nous pensons qu'ils auraient des comportements similaires. "
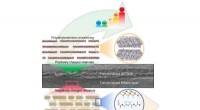
Toutes ces interactions ont été révélées en détail grâce à des modèles d'état de Markov qui analysent l'évolution des systèmes au fil du temps. Dans ce cas, les modèles ont révélé l'importance des sites secondaires qui soutiennent les ancres primaires du peptide. C'est là que la position 4 s'est imposée.
"Il y a les principaux, ancres canoniques que les gens connaissent, mais il y a ces interactions secondaires qui contribuent à la liaison et à la stabilité, " a déclaré Antunes. "Ce sont plus difficiles à capturer, mais dans cette étude, il semble que la position 4 joue un rôle très important. Lorsque vous le faites muter, il affecte le comportement du peptide lorsqu'il se délie de la molécule."
Les chercheurs ont modélisé les mutations du CMH pour voir comment elles influenceraient la liaison et ont découvert qu'elles soutenaient l'importance de la position 4 pour la stabilité du complexe.
Les cartes de potentiel électrostatique créées à partir de modèles par l'Université Rice montrent une protéine d'histocompatibilité majeure liée à un peptide tiré des virus du SRAS-CoV. De tels complexes dynamiques peuvent déclencher l'activation des cellules T qui font partie du système immunitaire. L'équipe Rice a découvert l'influence stabilisante d'un résidu de liaison alternatif (en position 4) commun au peptide étudié, en haut, et un associé au SARS-CoV-2, en bas, responsable de la maladie COVID-19. Crédit :Kavraki Lab/Université du riz
"Notre approche informatique a permis de faire des prédictions sur l'effet des mutations qui sont ensuite vérifiées expérimentalement, " a déclaré la co-auteur Cecilia Clementi, un ancien professeur Rice qui est récemment devenu professeur Einstein de physique à l'Université libre de Berlin.
Les chercheurs ont développé un processus en deux étapes pour simplifier la complexité informatique de l'analyse à l'échelle atomique de grosses molécules. La première étape a utilisé une technique appelée échantillonnage en parapluie pour accélérer l'exploration initiale des molécules. La deuxième, le stade exploratoire a utilisé l'échantillonnage adaptatif, dans lequel des simulations sont conduites pour accélérer la construction du modèle de Markov.
"Le défi est que ces MHC sont des systèmes assez grands pour les chimistes computationnels à simuler, " dit Abella, dont les recherches sur le sujet ont constitué une grande partie de sa thèse de doctorat. « Nous avons dû faire quelques approximations et tirer parti des avancées de ces classes de méthodes pour avancer.
"Nous ne sommes pas les premiers à étudier le déliement, mais ce qui caractérise notre travail par rapport aux autres, c'est que nous gardons la pleine résolution atomique dans nos simulations, " dit-il. " D'autres œuvres utilisent une technique connue sous le nom de chaîne de Markov Monte Carlo, alors que nous utilisons la dynamique moléculaire, ce qui nous permet d'incorporer le temps dans notre calcul pour capturer la cinétique."
Leurs méthodes peuvent être appliquées à d'autres complexes peptide-CMH avec des modèles 3-D existants. "C'était, en quelques sortes, une étude de faisabilité pour montrer que nous pouvons utiliser la dynamique moléculaire et construire un modèle d'état de Markov d'un système de cette taille, " dit Abella.
Les chercheurs ont également noté la pertinence de l'étude pour la lutte actuelle contre COVID-19, comme le peptide du SRAS qu'ils ont vu, QFKDNVILL, est très similaire au peptide NFKDQVILL dans le SARS-CoV-2, avec les mêmes pochettes de reliure aux positions 2, 4 et 9.
"Ces résultats suggèrent que les deux peptides peuvent se lier à HLA-A*24:02 et fournir des cibles pour les réponses antivirales des lymphocytes T, qui sont d'un grand intérêt au vu de la pandémie actuelle, " a déclaré le co-auteur Grégory Lizée, professeur au Département d'oncologie médicale du mélanome du MD Anderson. "Mais ces résultats mettent également en lumière de nombreuses autres cibles immunitaires potentielles, y compris ceux d'autres virus et même de cancers humains.
Kavraki a noté que le travail expérimental de la collaboratrice de longue date Lizée et Kyle Jackson, un assistant de recherche diplômé au laboratoire de Lizée qui a produit les protéines mutantes, étaient essentiels pour valider leurs simulations. Le propre laboratoire de Kavraki a remporté une subvention de recherche sur la réponse rapide de la National Science Foundation (NSF) pour aider à identifier des fragments de protéines virales du SRAS-CoV-2 comme cibles possibles pour le développement de vaccins.