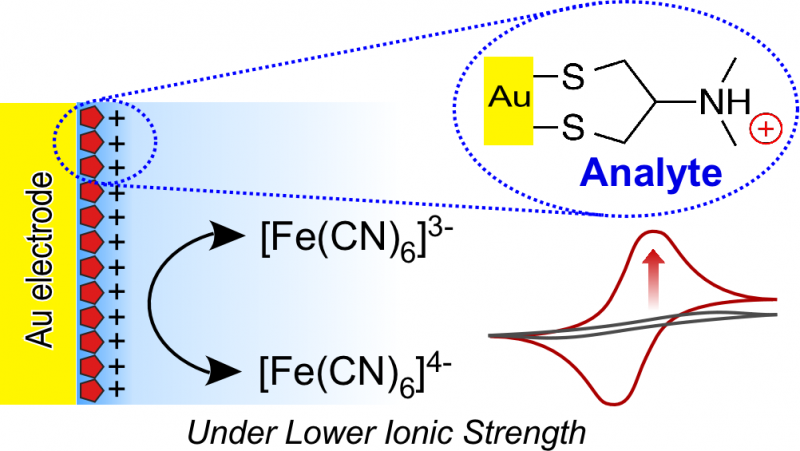
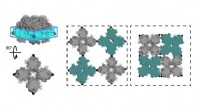
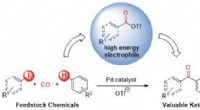
Figure 1 :Angle dièdre (l'angle formé par le plan créé par les atomes A, B, et C, et le plan créé par les atomes B, C, et D). Crédit :Fujitsu
Fujitsu Laboratories a annoncé aujourd'hui le développement d'une technologie de simulation moléculaire pour la découverte de médicaments qui peut estimer avec précision l'affinité de liaison, qui représente le degré auquel les protéines qui peuvent provoquer des maladies (protéines cibles) se lient à des substances chimiques qui pourraient devenir des médicaments candidats. Dans le processus de découverte de médicaments, il existe une demande pour une prédiction précise de l'affinité de liaison entre les protéines cibles et les substances chimiques, qui offre une estimation approximative de l'efficacité d'un médicament. La technologie de simulation moléculaire a été largement utilisée dans le passé comme méthode de prédiction de l'affinité de liaison, calculer les forces approximatives qui se produisent entre les atomes dans les molécules en utilisant la mécanique newtonienne. Le problème avec cette méthode, cependant, reste que le faible degré de précision de son estimation des paramètres les plus importants, le degré de torsion au niveau des sites de liaison. Cela signifie que la précision de son estimation de l'affinité de liaison globale est également médiocre.
Maintenant, Fujitsu Laboratories a développé une technologie de simulation moléculaire qui estime le degré de torsion d'une substance chimique, qui est directement lié à l'affinité de liaison prédite. La nouvelle technologie prend non seulement en compte l'emplacement de collage où la torsion se produira, mais aussi l'impact des atomes voisins. Les laboratoires Fujitsu ont évalué cette technologie pour 190 types de substances chimiques, comparer les résultats avec les résultats corrects obtenus à partir du premier calcul des principes, puis évaluer le taux d'erreur. En faisant cela, il a pu confirmer que le taux d'erreur dans l'estimation du degré de torsion était, en moyenne, un dixième de celui de la technologie précédente. Il est prévu que l'utilisation de cette nouvelle technologie dans la découverte de médicaments basée sur les TI, avec sa capacité à estimer avec précision l'affinité de liaison des protéines et substances chimiques ciblées, offre le potentiel d'efforts révolutionnaires de découverte de nouveaux médicaments qui ne pouvaient pas être atteints avec les approches précédentes.
La découverte de nouveaux médicaments nécessite des dépenses et des délais importants qui peuvent être mesurés en décennies, conduisant à une recherche mondiale de nouvelles méthodes de découverte de médicaments. L'une des méthodes qui a suscité un intérêt considérable est la découverte de médicaments basée sur les technologies de l'information, une nouvelle méthode de découverte de médicaments utilisant des ordinateurs qui permet de créer des substances chimiques en tant que candidats pour de nouveaux médicaments avec une forte probabilité de succès. La découverte de médicaments basée sur les TI est devenue un point focal pour les attentes en tant que technologie révolutionnaire pour la création de nouveaux médicaments, car contrairement aux méthodes précédentes d'essais et d'erreurs, dans lequel des substances chimiques sont créées et testées à plusieurs reprises, cette approche permet de concevoir virtuellement des substances chimiques et d'estimer leurs effets.
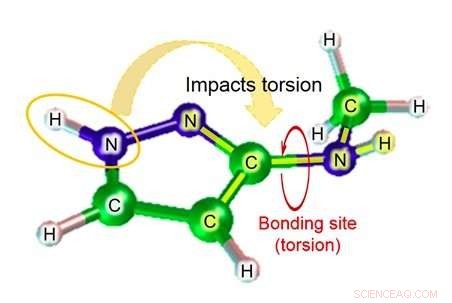
Figure 2 :Exemple de structure moléculaire :3-(méthylamino)pyrazole. Crédit :Fujitsu
Les effets d'une substance chimique en tant que médicament sont exprimés lorsque la substance chimique se lie à une protéine cible. Lorsque la substance chimique se lie à la protéine cible, elle peut changer de forme en fonction de celle de la protéine cible. Le degré de déformation, à savoir, les paramètres qui indiquent l'étendue de ce changement de forme, est directement lié à l'affinité de liaison de la substance et de la protéine, et donne une idée approximative de son effet en tant que médicament. Compte tenu de cela, il existe une forte demande pour la capacité de prédire avec précision cette valeur. Pour calculer le degré de déformation d'une substance chimique, il existe des méthodes basées sur la mécanique quantique et des méthodes basées sur la mécanique newtonienne. Le calcul des premiers principes basé sur la mécanique quantique permet des calculs extrêmement précis, résoudre les états des électrons à partir des types et des positions des atomes impliqués. D'autre part, cependant, la capacité des premiers principes à effectuer des calculs précis conduit nécessairement à un temps énorme requis pour effectuer les calculs. Afin de simuler le degré de déformation de nombreuses substances chimiques, le temps requis est de l'ordre des années, rendant cette méthode peu pratique. D'autre part, les calculs approximatifs basés sur des simulations moléculaires sont extrêmement rapides, en utilisant la mécanique newtonienne pour calculer les forces entre les atomes au sein des molécules, et peut même gérer assez facilement de grosses molécules comme des protéines. Par conséquent, cette méthode est largement utilisée. Avec la mécanique newtonienne, les forces entre les atomes s'expriment de la manière suivante :
Parmi ceux-ci, lorsqu'une substance chimique est liée à une protéine cible, le degré de torsion de la liaison représente le degré de déformation important. Avec la technologie existante, cependant, la précision de l'estimation du paramètre angle dièdre (figure 1), qui est nécessaire pour calculer le degré de torsion de la liaison, est assez faible, entraînant le problème de faible précision dans l'estimation de l'affinité de la liaison dans la simulation.
Les Laboratoires Fujitsu développent une technologie de simulation moléculaire depuis plus de dix ans. Maintenant, en utilisant les connaissances qu'il a acquises grâce à des efforts antérieurs, Fujitsu Laboratories a développé une technologie de simulation moléculaire qui peut estimer le paramètre d'angle dièdre en prenant en considération l'impact des atomes à proximité de la liaison. La technologie existante estime le paramètre d'angle dièdre sur la base d'un total de quatre atomes - les deux atomes de la liaison pertinente, et les autres atomes auxquels chacun de ces atomes était lié. Selon la structure de la molécule, cependant, il y a des cas où des atomes au-delà de ces quatre pourraient avoir un impact significatif, et dans ces cas, la marge d'erreur de l'estimation pourrait être assez grande. Avec cette technologie, Fujitsu Laboratories a créé une base de données de formules d'estimation pour les modèles de structure partielle où l'impact des atomes plus éloignés du site de liaison pourrait être important, ainsi que pour le degré de torsion des substances chimiques qui serait attendu dans ce cas. En utilisant la formule d'estimation pertinente pour trouver le degré de torsion (figure 2) dans le cas des molécules correspondant à la base de données pour les structures partielles, il est même devenu possible de faire des estimations très précises pour la torsion moléculaire, ce qui était auparavant difficile à calculer avec précision.
Lorsque Fujitsu Laboratories a intégré cette technologie dans le logiciel qu'il avait développé pour générer des paramètres sophistiqués pour les forces entre atomes (FF-FOM), il a pu confirmer que les résultats étaient conformes à des calculs précis.

Figure 3 :Évaluation de la performance des valeurs des paramètres d'angle dièdre à l'aide de 190 types de structures de composés chimiques. Crédit :Fujitsu
Lorsque Fujitsu Laboratories a évalué la différence entre les résultats de cette technologie et les résultats d'un calcul à partir des premiers principes pour l'estimation du degré de torsion avec 190 types de substances chimiques, il était inférieur au dixième de celui de la technologie précédente, en moyenne, 0,6 kcal/mol en dessous des fluctuations thermiques de la température ambiante, confirmant que la nouvelle technologie est pratique. Parce qu'il peut estimer avec précision l'affinité de liaison des protéines cibles et des substances chimiques, on s'attend à ce que l'utilisation de cette technologie conduise à la création de nouveaux médicaments révolutionnaires grâce à son utilisation dans la découverte de médicaments basée sur les technologies de l'information.