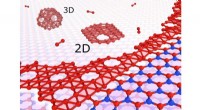
Les nanostructures de carbone pourraient devenir plus faciles à concevoir et à synthétiser grâce à une méthode d’apprentissage automatique qui prédit leur croissance sur les surfaces métalliques. La nouvelle approche, développée par des chercheurs de l'Université japonaise du Tohoku et de l'Université Jiao Tong de Shanghai en Chine, facilitera l'exploitation de la polyvalence chimique unique de la nanotechnologie du carbone. La méthode a été publiée dans la revue Nature Communications .
La croissance de nanostructures de carbone sur diverses surfaces, notamment sous forme de films atomiquement minces, a été largement étudiée, mais on sait peu de choses sur la dynamique et les facteurs au niveau atomique régissant la qualité des matériaux résultants. "Nos travaux répondent à un défi crucial pour réaliser le potentiel des nanostructures de carbone dans les dispositifs électroniques ou de traitement de l'énergie", déclare Hao Li de l'équipe de l'Université de Tohoku.
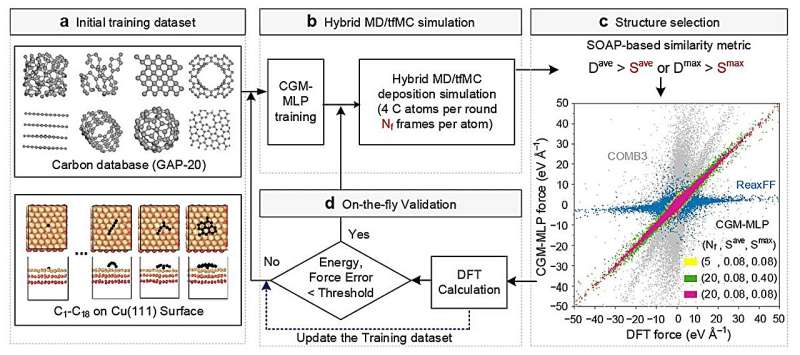
Le large éventail de surfaces possibles et la sensibilité du processus à plusieurs variables rendent l’investigation expérimentale directe difficile. Les chercheurs se sont donc tournés vers les simulations d'apprentissage automatique comme moyen plus efficace d'explorer ces systèmes.
Grâce à l’apprentissage automatique, divers modèles théoriques peuvent être combinés avec des données issues d’expériences chimiques pour prédire la dynamique de la croissance cristalline du carbone et déterminer comment elle peut être contrôlée pour obtenir des résultats spécifiques. Le programme de simulation explore les stratégies et identifie celles qui fonctionnent et celles qui ne fonctionnent pas, sans qu'il soit nécessaire que des humains guident chaque étape du processus.
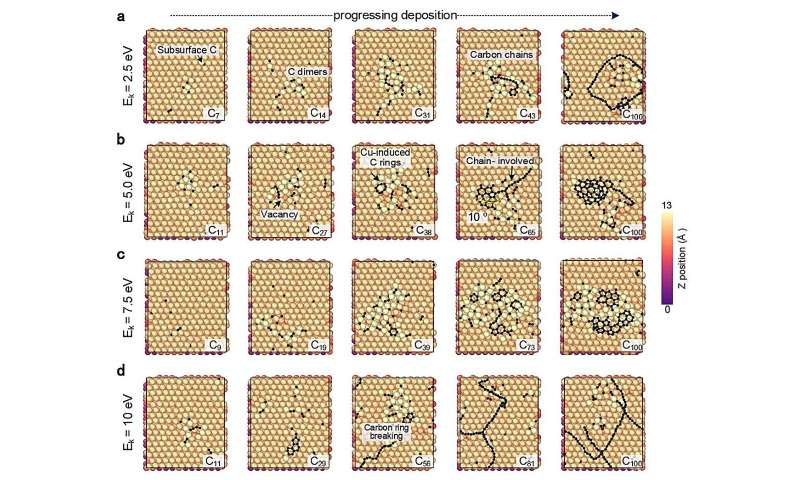
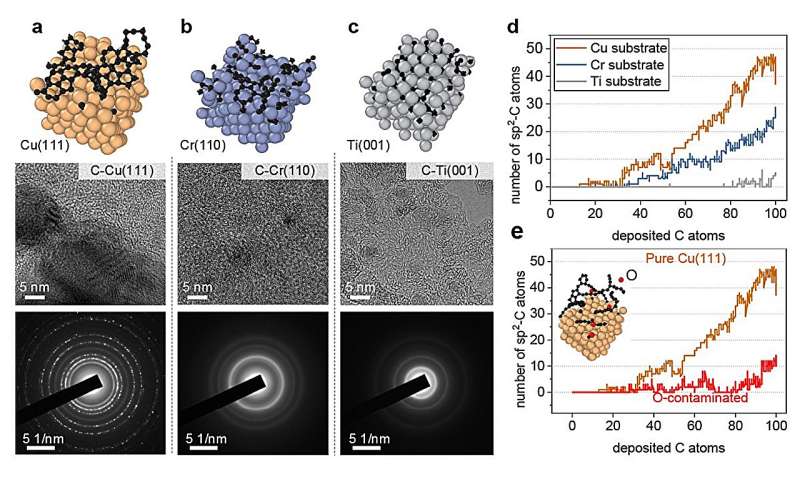
Les chercheurs ont testé cette approche en étudiant des simulations de la croissance du graphène, une forme de carbone, sur une surface de cuivre. Après avoir établi le cadre de base, ils ont montré comment leur approche pouvait également être appliquée à d'autres surfaces métalliques, telles que le titane, le chrome et le cuivre contaminés par l'oxygène.
La répartition des électrons autour des noyaux des atomes dans différentes formes de cristaux de graphène peut varier. Ces différences subtiles dans la structure atomique et la disposition électronique affectent les propriétés chimiques et électrochimiques globales du matériau. L'approche d'apprentissage automatique peut tester comment ces différences affectent la diffusion des atomes individuels et des atomes liés et la formation de chaînes, d'arcs et de structures annulaires carbonées.
L’équipe a validé les résultats des simulations par des expériences et a constaté qu’ils correspondaient étroitement. "Dans l'ensemble, notre travail fournit une méthode pratique et efficace pour concevoir des substrats métalliques ou en alliages afin d'obtenir les nanostructures de carbone souhaitées et d'explorer d'autres opportunités", explique Li.
Il ajoute que les travaux futurs s'appuieront sur ces données pour étudier des sujets tels que les interfaces entre solides et liquides dans les catalyseurs avancés et les propriétés chimiques des matériaux utilisés pour le traitement et le stockage de l'énergie.
Plus d'informations : Di Zhang et al, Modèle d'apprentissage automatique actif pour la simulation dynamique et les mécanismes de croissance du carbone sur une surface métallique, Nature Communications (2024). DOI :10.1038/s41467-023-44525-z
Fourni par l'Université du Tohoku