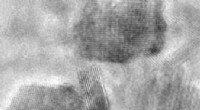
Représentation schématique des différents termes énergétiques contribuant à l'énergie d'adsorption, et la différence de densité de charge de 2H-P après adsorption sur Cu (111) à une séparation de 12,8 Angstrom. Crédit :M. Müller/TU Munich
Alors que nous continuons à réduire les composants électroniques, les méthodes de fabrication descendantes commencent à approcher une limite physique à l'échelle nanométrique. Plutôt que de continuer à rogner sur cette limite, une solution intéressante consiste à utiliser l'auto-assemblage ascendant de blocs de construction moléculaires pour construire des dispositifs à l'échelle nanométrique.
L'auto-assemblage réussi est une danse minutieusement chorégraphiée, dans lequel les forces attractives et répulsives au sein des molécules, entre chaque molécule et ses voisines, et entre les molécules et la surface qui les supporte, doivent tous être pris en compte. Pour mieux comprendre le processus d'auto-assemblage, des chercheurs de l'Université technique de Munich ont caractérisé les contributions de toutes les composantes d'interaction, telles que la liaison covalente et les interactions de van der Waals entre les molécules et entre les molécules et une surface.
« Dans un cas idéal, le plus petit appareil possible a la taille d'un seul atome ou molécule, " a déclaré Katharina Diller, qui a travaillé comme chercheur postdoctoral dans le groupe de Karsten Reuter à l'Université technique de Munich. Reuters et ses collègues présentent leur travail cette semaine en Le Journal de Physique Chimique .
Un tel exemple est un commutateur à porphyrine unique, qui occupe une surface de seulement un nanomètre carré. La molécule de porphine, qui a fait l'objet de cette étude, est encore plus petit que cela. Les porphyrines sont un groupe de composés chimiques annelés qui comprennent notamment l'hème - responsable du transport de l'oxygène et du dioxyde de carbone dans le sang - et la chlorophylle. Dans les applications dérivées de la synthèse, les porphyrines sont étudiées pour leurs utilisations potentielles comme capteurs, colorants photosensibles dans les cellules solaires organiques, et des aimants moléculaires.
Les chercheurs de TU Munich ont évalué les interactions de la molécule de porphyrine 2H-porphine en utilisant la théorie fonctionnelle de la densité, une méthode de modélisation informatique quantique utilisée pour décrire les propriétés électroniques des molécules et des matériaux. Leurs simulations ont été réalisées sur le supercalculateur hautes performances SuperMUC du Leibniz-Rechenzentrum à Garching.
Les substrats métalliques sur lesquels les chercheurs ont choisi d'assembler les molécules de porphyrine, les surfaces monocristallines serrées de cuivre et d'argent, sont largement utilisés comme substrats dans la science des surfaces. Cela est dû à la nature dense des surfaces, qui permettent aux molécules de présenter un environnement d'adsorption lisse. En outre, le cuivre et l'argent réagissent chacun différemment avec les porhyrines - la molécule s'adsorbe plus fortement sur le cuivre, tandis que l'argent fait un meilleur travail pour garder la structure électronique de la molécule intacte - permettant aux chercheurs de surveiller une variété d'effets concurrents pour des applications futures.
Dans leur simulation, des molécules de porphyrine ont été placées sur une plaque de cuivre ou d'argent, qui a été répété périodiquement pour simuler une surface étendue. Après avoir trouvé la géométrie optimale dans laquelle les molécules s'adsorberaient à la surface, les chercheurs ont modifié la taille de la plaque métallique pour augmenter ou diminuer la distance entre les molécules, simulant ainsi différentes couvertures moléculaires. La configuration informatique leur a donné un interrupteur pour activer et désactiver les contributions énergétiques des molécules voisines, afin d'observer le jeu des interactions individuelles.
Diller et Reuters, avec ses collègues Reinhard Maurer et Moritz Müller, qui est le premier auteur de l'article, ont constaté que les faibles interactions de van der Waals à longue distance ont donné la plus grande contribution à l'interaction molécule-surface, et a montré que les méthodes souvent employées pour quantifier les charges électroniques dans le système doivent être utilisées avec prudence. Étonnamment, alors que les interactions directes entre les molécules sont négligeables, le chercheur a trouvé des indications pour des interactions molécule-molécule médiées par la surface à des couvertures moléculaires plus élevées.
"L'analyse de la structure électronique et des composants individuels d'interaction nous permet de mieux comprendre l'auto-assemblage de la porphine adsorbée sur le cuivre et l'argent, et permet en outre des prédictions pour des analogues de porphyrine plus complexes, " Diller dit. " Ces conclusions, cependant, viennent sans encore considérer les effets du mouvement atomique à température finie, que nous n'avons pas étudié dans cet ouvrage."