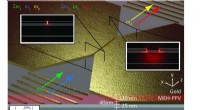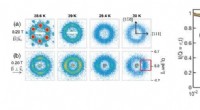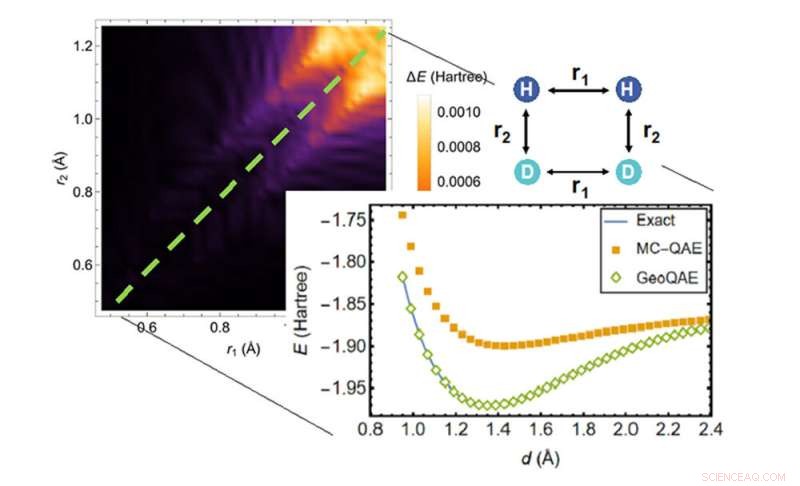
Dans le calcul de la surface d'énergie potentielle de la réaction chimique de H2;+ D2 → 2HD, le nouvel algorithme (losanges verts) surpasse l'algorithme précédent (carrés oranges) pour trouver la solution la plus précise (ligne bleue). Crédit :Laboratoire national de Brookhaven
Une équipe de chercheurs du Laboratoire national de Brookhaven du Département américain de l'énergie (DOE) et de l'Université de Stony Brook ont mis au point un nouvel algorithme quantique pour calculer les énergies les plus basses des molécules à des configurations spécifiques lors de réactions chimiques, y compris lorsque leurs liaisons chimiques sont rompues. Comme décrit dans Recherche d'examen physique , par rapport à des algorithmes existants similaires, y compris la méthode précédente de l'équipe, le nouvel algorithme améliorera considérablement la capacité des scientifiques à calculer avec précision et fiabilité la surface d'énergie potentielle dans les molécules en réaction.
Pour ce travail, Deyu Lu, physicien du Center for Functional Nanomaterials (CFN) au Brookhaven Lab, a travaillé avec Tzu-Chieh Wei, professeur agrégé spécialisé en science de l'information quantique au C.N. Yang Institute for Theoretical Physics de l'Université Stony Brook, Qin Wu, théoricien au CFN, et Hongye Yu, titulaire d'un doctorat. étudiant à Stony Brook.
"Comprendre la mécanique quantique d'une molécule, son comportement au niveau atomique, peut fournir des informations essentielles sur ses propriétés chimiques, telles que sa stabilité et sa réactivité", a déclaré Lu.
Une propriété particulière qu'il a été difficile de déterminer est l'état fondamental d'une molécule :le point où l'énergie électronique totale de la molécule (y compris l'énergie cinétique et potentielle) est à son plus bas et où rien en dehors de ce « système moléculaire » n'excite ou ne charge l'énergie de la molécule. électrons. Lorsque la structure atomique d'un système chimique devient plus complexe, comme dans une grosse molécule, beaucoup plus d'électrons peuvent interagir. Ces interactions rendent extrêmement difficile le calcul de l'état fondamental de molécules complexes.
Le nouvel algorithme quantique améliore l'algorithme précédent pour résoudre ce problème de manière créative. Il exploite une déformation géométrique lisse réalisée en faisant varier en continu les longueurs de liaison ou les angles de liaison dans la structure de la molécule. Avec cette approche, les scientifiques disent qu'ils peuvent calculer l'état fondamental des molécules de manière très précise, même lorsque les liaisons chimiques se rompent et se reforment au cours des réactions chimiques.
Construire le terrain
"Lorsqu'il s'appuie uniquement sur des méthodes informatiques traditionnelles, ce problème d'état fondamental contient trop de variables à résoudre, même sur les supercalculateurs les plus puissants", a déclaré Lu.
Vous pouvez considérer un algorithme comme un ensemble d'étapes pour résoudre un problème particulier. Les ordinateurs classiques peuvent exécuter des algorithmes complexes, mais à mesure qu'ils deviennent plus gros et plus impliqués, ils peuvent devenir trop difficiles ou prendre trop de temps pour que les ordinateurs classiques puissent les résoudre de manière réaliste. Les ordinateurs quantiques peuvent accélérer le processus en tirant parti des règles de la mécanique quantique.
En informatique classique, les données sont stockées dans des bits qui ont une valeur de 1 ou 0. Un bit quantique, appelé qubit, peut avoir une valeur au-delà de 0 ou 1, il peut même avoir une valeur de 0 et 1, dans un soi-disant superposition quantique. En principe, ces qubits plus "flexibles" peuvent stocker une plus grande quantité d'informations que les bits classiques. Si les scientifiques peuvent trouver des moyens d'exploiter la capacité de transport d'informations des qubits, la puissance de calcul peut augmenter de façon exponentielle avec chaque qubit supplémentaire.
Les Qubits, cependant, sont assez fragiles. Ils peuvent souvent tomber en panne lors de l'extraction des informations. Lorsqu'un dispositif quantique interagit avec l'environnement qui l'entoure, il peut générer du bruit ou des interférences qui détruisent l'état quantique. Les changements de température, les vibrations, les interférences électromagnétiques et même les défauts matériels peuvent également entraîner la perte d'informations sur les qubits.
Pour pallier ces écueils, les scientifiques ont développé une solution hybride qui tire parti des deux algorithmes de calcul classiques, plus stables et pratiques.
Lu et Wei ont commencé leurs recherches sur les approches hybrides d'informatique classique et quantique en 2019. Cette subvention annuelle promeut la collaboration entre le Laboratoire national de Brookhaven et l'Université de Stony Brook en finançant des initiatives de recherche conjointes qui s'alignent sur les missions des deux institutions. Avec ce travail initial, Lu et Wei se sont d'abord concentrés sur la résolution du problème de l'état fondamental en remplaçant les algorithmes classiques les plus "coûteux" - ceux qui étaient beaucoup plus complexes et nécessitaient beaucoup plus d'étapes (et plus de temps de calcul) pour être complétés - par des algorithmes quantiques. .
Renforcer les liens, créer de nouveaux chemins
Les chercheurs notent que les algorithmes quantiques existants présentent tous des inconvénients pour résoudre le problème de l'état fondamental, y compris celui développé par Wei et Yu en 2019. Alors que certains algorithmes populaires sont précis lorsqu'une molécule est à sa géométrie d'équilibre - son arrangement naturel d'atomes en trois dimensions - ces algorithmes peuvent devenir peu fiables lorsque les liaisons chimiques sont rompues à de grandes distances atomiques. La formation et la dissociation des liaisons jouent un rôle dans de nombreuses applications, telles que la prédiction de la quantité d'énergie nécessaire pour déclencher une réaction chimique. Les scientifiques avaient donc besoin d'un moyen de résoudre ce problème lorsque les molécules réagissent. Ils avaient besoin de nouveaux algorithmes quantiques capables de décrire la rupture de liaison.
Pour cette nouvelle version de l'algorithme, l'équipe a travaillé avec le Brookhaven-Lab-led Co-design Center for Quantum Advantage (C2QA), qui a été formé en 2020. Wei contribue à la poussée logicielle du centre, qui se spécialise dans les algorithmes quantiques. Le nouvel algorithme de l'équipe utilise une approche adiabatique, qui effectue des changements graduels, mais avec quelques adaptations qui garantissent qu'il reste fiable lorsque les liaisons chimiques sont rompues.
"Un processus adiabatique fonctionne en adaptant progressivement les conditions d'un système mécanique quantique", a expliqué Lu. "D'une certaine manière, vous atteignez une solution en très petites étapes. Vous faites évoluer le système d'un modèle simple et soluble à la cible finale, généralement un modèle plus difficile. En plus de l'état fondamental, cependant, un système à plusieurs électrons a de nombreux états excités à des énergies plus élevées. Ces états excités peuvent poser un défi lors de l'utilisation de cette méthode pour calculer l'état fondamental."
Wei a comparé un algorithme adiabatique à la conduite sur une autoroute, "si vous voyagez d'une ville à l'autre, il y a plusieurs chemins pour y arriver, mais vous voulez trouver le plus sûr et le plus efficace."
Dans le cas de la chimie quantique, la clé est de trouver un "écart d'énergie" suffisamment grand entre l'état fondamental et les états excités où aucun état électronique n'existe. Avec un écart suffisamment grand, les véhicules de la métaphore de l'autoroute ne "traverseront pas les voies", afin que leurs trajectoires puissent être tracées avec précision.
"Un grand écart signifie que vous pouvez aller plus vite, donc, dans un sens, vous essayez de trouver une autoroute moins encombrée pour rouler plus vite sans rien heurter", a déclaré Wei.
"Avec ces algorithmes, l'entrée du chemin est une solution simple et bien définie de l'informatique classique", a noté Wei. "Nous savons également où se trouve la sortie - l'état fondamental de la molécule - et nous essayions de trouver un moyen de la connecter à l'entrée de la manière la plus naturelle, une ligne droite.
"Nous l'avons fait dans notre premier article, mais la ligne droite avait des obstacles causés par la réduction de l'écart énergétique et le croisement des chemins. Nous avons maintenant une meilleure solution."
Lorsque les scientifiques ont testé l'algorithme, ils ont démontré que même avec des changements de longueur de liaison finis, la version améliorée fonctionnait toujours avec précision pour l'état fondamental.
"Nous sommes allés au-delà de notre zone de confort, car la chimie n'est pas notre objectif", a déclaré Wei. "Mais c'était bien de trouver une application comme celle-ci et de favoriser ce type de collaboration avec le CFN. Il est important d'avoir des perspectives différentes en matière de recherche."
Il a noté l'effort accumulé de nombreuses personnes. "Dans le grand schéma, je pense que nous apportons une petite contribution, mais cela pourrait être une base pour d'autres travaux dans ces domaines", a-t-il déclaré. "Cette recherche n'est pas seulement fondamentale, mais une excellente illustration de la façon dont différentes institutions et installations peuvent s'unir pour tirer parti de leurs domaines d'expertise." Vers un ordinateur quantique qui calcule l'énergie moléculaire