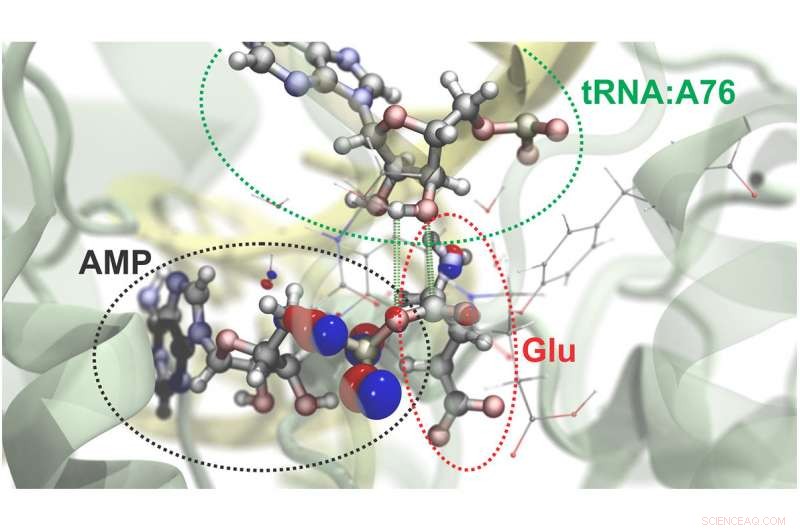
Les chercheurs peuvent simuler la dynamique atomique et subatomique dans de grands systèmes moléculaires. Voici une visualisation du processus par lequel le glutamate d'acide aminé (Glu) est attaché à une région spécifique de son ARN de transfert (ARNt). Une molécule riche en énergie, ATP, entraîne cette réaction et est converti en AMP dans le processus. Les bulles rouges et bleues représentent la probabilité de trouver des électrons dans des régions particulières. Les lignes pointillées vertes délimitent les atomes qui se lient dans cette réaction chimique. Crédit :Rafael Bernardi, Zan Luthey-Schulten et Marcelo Melo
Les scientifiques ont construit un "microscope informatique" qui peut simuler les forces atomiques et subatomiques qui entraînent les interactions moléculaires. Cet outil rationalisera les efforts pour comprendre la chimie de la vie, modéliser de grands systèmes moléculaires et développer de nouveaux agents pharmaceutiques et industriels, disent les chercheurs.
Ils rapportent leurs découvertes dans le journal Méthodes naturelles .
Les scientifiques ont combiné deux approches informatiques utilisées pour simuler les interactions moléculaires. La première, un programme de dynamique moléculaire à l'échelle nanométrique connu sous le nom de NAMD, utilise des méthodes de mécanique classique pour modéliser la structure et simuler le comportement de centaines de millions d'atomes individuels. Le deuxième programme zoome sur le domaine subatomique, simuler les interactions des protons, neutrons et électrons. La modélisation à cette échelle de la mécanique quantique demande beaucoup de puissance de calcul, les chercheurs ont donc mis en œuvre une méthode pour diviser les grosses molécules en régions de mécanique classique et quantique. Cela leur permet de concentrer leurs ressources de calcul sur de petites régions impliquées dans des interactions critiques, comme la création ou la rupture de liaisons chimiques.
Les programmes de mécanique moléculaire et de mécanique quantique sont disponibles depuis des années, et d'autres équipes ont travaillé pour les combiner, a déclaré le professeur de chimie de l'Université de l'Illinois, Zaida (Zan) Luthey-Schulten, qui a dirigé la nouvelle recherche avec son mari, U. of I. professeur de physique Klaus Schulten. Mais le nouvel effort rationalise le processus de mise en place, effectuer et analyser les simulations.
"Nous l'avons mis en place pour que les chercheurs puissent facilement choisir comment ils vont partitionner leurs propres systèmes, " Luthey-Schulten a déclaré. "Mes propres étudiants l'essaient, et la plupart d'entre eux sont capables de le faire sans trop de difficultés."
Schulten a développé le NAMD dans l'Illinois en 1995, en le combinant avec un logiciel de visualisation, VMD, qui permet aux chercheurs d'observer le déroulement des interactions moléculaires à grande échelle. Schulten, décédé en 2016, assimilé cette approche à la « construction d'un microscope informatique ».
Le microscope informatique est idéal pour modéliser les caractéristiques structurelles et les mouvements de grands complexes. Par exemple, en 2013, Schulten et ses collègues ont utilisé le NAMD pour modéliser la capside du VIH, qui est composé de plus de 1, 300 protéines identiques qui s'assemblent dans une structure semblable à une cage qui protège le virus jusqu'à ce qu'il pénètre dans une cellule hôte. Cette simulation a pris en compte les interactions de plus de 64 millions d'atomes et a nécessité l'utilisation du supercalculateur Blue Waters du National Center for Supercomputing Applications de l'U. of I. La nouvelle étude a également utilisé Blue Waters, cette fois pour améliorer la résolution du microscope informatique.

De gauche, étudiant diplômé Marcelo Melo, professeur de chimie Zaida Luthey-Schulten, Le chercheur postdoctoral Rafael Bernardi et ses collègues ont développé une nouvelle approche pour modéliser les grandes interactions moléculaires aux échelles atomique et subatomique. Leur travail rationalise la méthode pour d'autres scientifiques et étudiants. Crédit :L. Brian Stauffer
Le logiciel NAMD est conçu pour décrire le comportement des atomes individuels. Mais les atomes individuels impliqués dans des interactions et des réactions chimiques spécifiques ne se comportent pas toujours comme leurs homologues ailleurs. Comprendre comment elles varient nécessite d'examiner de plus près les forces subatomiques en jeu. Ceci est particulièrement important dans les régions dynamiques des molécules - par exemple, ces endroits où les liaisons chimiques sont faites ou rompues, les chercheurs ont dit.
Dans la nouvelle étude, l'équipe de recherche de l'Illinois s'est associée aux experts en QM Frank Neese, de l'Institut Max Planck de recherche sur le charbon à Mulheim an der Ruhr, Allemagne; et Gerd B. Rocha, de l'Université fédérale de Paraiba, à João Pessoa, Brésil.
Comme démonstration de la nouvelle approche, les chercheurs ont simulé le comportement chimique des ARN de transfert, molécules qui jouent un rôle clé dans la traduction de l'information génétique en protéines. En utilisant le NAMD, ils ont modélisé la structure moléculaire globale de l'ARNt au moment où une protéine spéciale charge un acide aminé dans l'ARNt. Ils ont divisé deux sites du complexe en régions nécessitant une approche plus ciblée de la mécanique quantique. (Regardez un film de la simulation.)
Les simulations subatomiques des interactions des deux régions ont permis à l'équipe d'exécuter des simulations de quatre scénarios différents qui permettraient à l'ARNt de fonctionner comme il le fait dans la cellule. Leurs simulations ont révélé que l'une des quatre voies chimiques potentielles était plus énergétiquement favorable que les autres et donc plus susceptible de se produire.
Les chercheurs ont également utilisé diverses méthodes pour partitionner le complexe d'ARNt entre les régions MM et QM et ont rendu compte de chaque approche.
« Nous n'avons pas choisi une seule voie ; nous en avons choisi autant que possible. Nous laissons la liberté à l'utilisateur. La façon dont vous la structurez dépend vraiment du système particulier que vous étudiez, " a déclaré Rafael Bernardi, chercheur postdoctoral à l'U. of I., un co-auteur principal de l'étude avec l'étudiant diplômé Marcelo Melo.
"Nous ne faisons pas tout le système quantique mécaniquement parce que cela prendrait une éternité à calculer, " dit Mélo.
"Le NAMD a été conçu - et c'était la vision de mon mari - pour traiter de très gros systèmes, " a déclaré Luthey-Schulten. " Maintenant, nous pouvons ajouter l'échelle subatomique à cela, ouvrant de vastes nouvelles possibilités de recherche. »