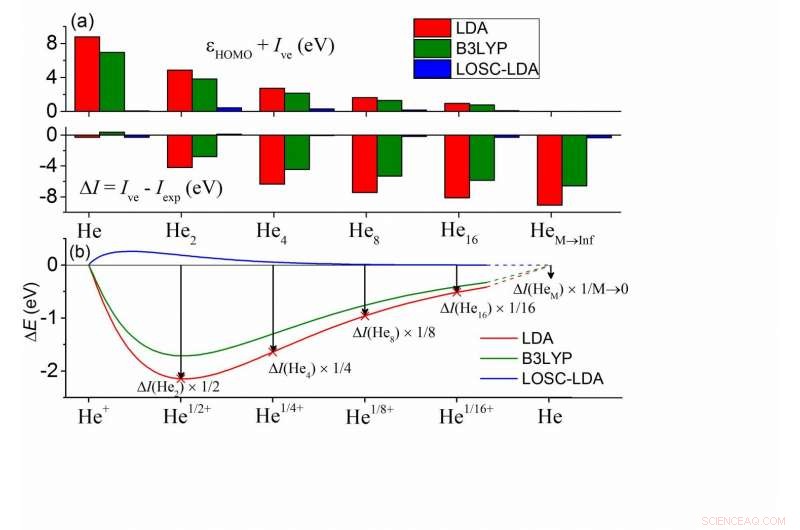
(a) Écarts entre les ?HOMO et -Ive calculés et entre Ive et Iexp pour une série de clusters HeM. Dans chaque amas, tous les atomes d'He sont chimiquement équivalents. Les atomes voisins les plus proches sont séparés de 10Å, et le Iexp de HeM est bien approximé par Iexp d'un atome de He. (b) Calcul de l'écart d'énergie totale par rapport à la condition de linéarité d'un atome d'He à charge fractionnelle en fonction de la charge fractionnaire . Ici ΔE(Heδ+) =E(Heδ+)-δE(He+)-(1-δ)E(He), et les valeurs de ont été mises à l'échelle dans la figure pour une comparaison directe avec (a). Crédit :©Science China Press
La théorie fonctionnelle de la densité de Kohn-Sham est l'une des théories les plus réussies en chimie. Il est formellement rigoureux; son coût de calcul relativement faible et sa précision compétitive dans les systèmes de petite et moyenne taille en font l'une des méthodes les plus populaires dans les calculs de structure électronique, et peut-être le seul choix pour modéliser les effets quantiques des électrons dans les grands systèmes chimiques et biologiques. Néanmoins, la fonctionnelle exacte n'est pas explicitement connue et les approximations fonctionnelles de densité (DFA) de pointe souffrent d'erreurs systématiques. L'une des erreurs dominantes dans les DFA est l'erreur de délocalisation, qui est omniprésent et se manifeste de diverses manières. Cela a été un problème ouvert difficile pendant des décennies. Récemment, Chen Li, Neil Qiang Su et Weitao Yang de l'Université Duke et Xiao Zheng de l'Université des sciences et technologies de Chine ont développé un nouveau cadre de correction d'échelle orbitale localisée (LOSC) qui démontre l'élimination systématique de l'erreur de délocalisation.
Les conséquences majeures des erreurs de délocalisation dans les DFA peuvent être classées en deux aspects :(1) erreur d'énergie orbitale Kohn-Sham (KS) et (2) erreur d'énergie totale. L'erreur de délocalisation conduit à des échecs importants dans les applications pratiques telles que la sous-estimation non physique des bandes interdites, les distributions d'électrons excessivement délocalisées et les mauvais transferts de charges. Concernant ces deux aspects des erreurs, les deux se manifestent de manière dépendante de la taille. En particulier, parmi les énergies orbitales KS, l'énergie orbitale moléculaire occupée (HOMO) la plus élevée, HOMO, est supposé être en accord avec le potentiel d'ionisation vertical (négatif) (-Ive), comme requis par le fonctionnel exact. Par ailleurs, les Ive calculés par les DFA sont censés être en accord avec la valeur expérimentale, Iexp.
Pour les DFA couramment utilisés, les deux conditions ne sont pas remplies. Comme le montrent les calculs d'amas d'hélium dans la sous-figure (a), l'énergie HOMO par approximation de la densité locale (LDA) montre une erreur positive par rapport à -Ive alors que Ive présente une erreur négative, et les deux erreurs sont affichées en fonction de la taille, suggérant que l'erreur de délocalisation des DFA doit apparaître d'une manière ou d'une autre, ou les deux, tandis que la somme des grandeurs reste inchangée. De plus, ceci est répandu dans toutes sortes de DFA, y compris le B3LYP fonctionnel le plus populaire, comme indiqué en vert.
Pour comprendre ces erreurs, les chimistes peuvent les cartographier dans les erreurs d'un atome d'hélium chargé en fraction, comme illustré dans la sous-figure (b). Les énergies du système fractionnaire sont censées évoluer linéairement avec le nombre d'électrons fractionnaires n (0?n <1) pour la fonctionnelle exacte. La sous-figure (b) montre l'écart d'énergie par rapport à la condition de linéarité pour un seul atome d'He, où les énergies du système fractionnaire sont sous-estimées - cela définit l'erreur de délocalisation. De plus, l'erreur de charge fractionnaire dans (b) a une correspondance 1-1 avec l'erreur d'énergie totale ΔI =Ive - Iexp. Dans la limite de M tend vers l'infini, on peut en déduire que ΔI est d'accord avec l'erreur de pente de la courbe E(N) à l'entier, qui est exactement l'erreur d'énergie HOMO d'un seul atome d'He. Par conséquent, toutes les erreurs sont interconnectées. Il est ainsi possible d'éliminer systématiquement l'erreur de délocalisation en (1) supprimant les erreurs en (b) et (2) en garantissant une suppression cohérente en taille de l'erreur dans tous les systèmes.
Dans le présent travail du LOSC, les auteurs ont inventé de nouvelles variables locales, appelés orbitallettes, qui sont des orbitales localisées (LO) qui atteignent la localité dans les espaces spatiaux et énergétiques. A travers ces orbitales, ils ont pu exprimer la densité électronique et la matrice de densité KS dans la représentation locale, où la matrice d'occupation locale composée de nombres fractionnaires apparaît naturellement. Par conséquent, ces variables locales sont capables de capturer des informations fractionnaires locales de manière précise et complète dans l'espace orbital, et servent de blocs de construction parfaits pour la fonction LOSC. Par ailleurs, en réécrivant les termes des fonctionnelles GSC et LSC en termes de formules de correction de courbure et en faisant analogie avec le LOSC, ils ont introduit une matrice de courbure locale, dont les éléments sont conçus comme une fonctionnelle des orbitallettes, être en correspondance un à un avec la matrice d'occupation locale. Finalement, la formule LOSC est écrite comme une expression explicite et élégante sur la fonctionnelle parente en fonction de ces deux matrices.
Le LOSC réalise toutes les caractéristiques souhaitables. Dans la figure ci-dessus, les erreurs LOSC-LDA sont essentiellement négligeables, indiquant la validité et la cohérence de la taille de la correction. Celles-ci ont également été validées par les courbes de dissociation bien améliorées des cations moléculaires diatomiques, allant de H2+, He2+, le cation dimère d'eau et le cation dimère de benzène. L'HOMO, Les erreurs d'énergie LUMO ont été considérablement réduites et les bonnes performances se maintiennent à mesure que la taille du système augmente. Ceci a été démontré par des tests sur des oligomères polyacène et trans-polyacétylène. Par ailleurs, le LOSC est capable de récupérer la bonne densité électronique lorsque les DFA parents la rendent qualitativement erronée, comme le montre l'exemple d'un anion chlore solvaté lorsqu'il est privé d'un électron.
Les performances de LOSC semblent donc prometteuses d'éliminer systématiquement l'erreur de délocalisation au sein des fonctionnelles de densité KS. Il convient de noter que contrairement au paradigme traditionnel de conception de fonctionnelles utilisant la densité, les gradients de densité, la densité d'énergie cinétique, etc., le cadre LOSC implique des ingrédients complètement nouveaux, les orbitales, qui sont elles-mêmes des fonctionnelles implicites de la matrice densité KS, et ont démontré leur capacité unique à résoudre les problèmes de longue date associés aux AFD traditionnels. Cela reflète un changement de paradigme dans la conception des fonctionnelles, et élargit considérablement la voie dans l'exploration de la fonctionnelle exacte dans son propre espace de vie. Dans ce sens, Le LOSC a inauguré la tendance au développement d'une nouvelle génération d'approximations fonctionnelles de densité, promouvoir la théorie de la fonctionnelle de la densité à un nouveau niveau de précision.