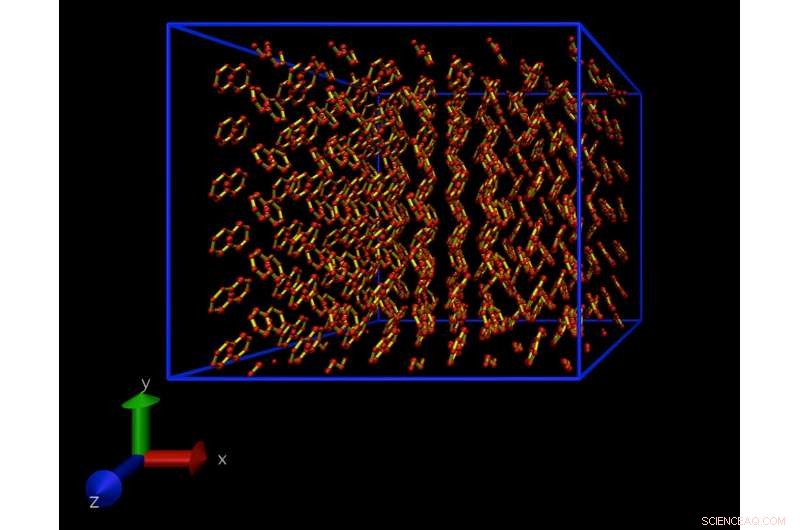
Un instantané d'une simulation de dynamique moléculaire d'un modèle atomistique d'un cristal de naphtalène. Ce cristal se répète périodiquement dans toutes les directions, pour éliminer les effets de surface. Crédit :Daan Frenkel, Université de Cambridge
La solubilité d'une substance donnée - la mesure de la façon dont la substance se dissout dans une autre substance appelée solvant - dépend de propriétés de base telles que la température et la pression, ainsi que les identités chimiques de la substance dissoute (le soluté) et du solvant.
Prédire la solubilité est important pour une variété d'applications. Dans le domaine pharmaceutique, par exemple, il est crucial de connaître la solubilité d'un médicament car elle détermine directement sa disponibilité pour l'organisme. L'industrie pétrolière fournit un autre exemple :les substances à faible solubilité peuvent former des écailles ou des dépôts indésirables dans les tuyaux ou sur les foreuses, causant des blocages et d'autres gros problèmes.
Malgré l'importance de prédire la solubilité, ce n'est pas une affaire facile. Une approche, en utilisant des simulations "force brute", nécessite des temps de calcul longs. D'autres techniques, tout en étant plus rapide, ne parviennent pas à prédire des valeurs de solubilité précises. Cette semaine à Le Journal de Physique Chimique , les chercheurs rapportent un nouveau type de logiciel qui permet des estimations pratiques de la solubilité de pratiquement n'importe quelle substance moléculaire sur de larges plages de température et de pression. Le code utilise des logiciels open source facilement disponibles et devrait être largement adopté.
Daan Frenkel de l'Université de Cambridge au Royaume-Uni a travaillé avec ses collègues Lunna Li, aussi à Cambridge, et Tim Totton, de British Petroleum, pour développer le code.
"Nous avons fait le choix conscient d'utiliser des logiciel disponible gratuitement car nous voulions rendre notre approche accessible à tous, " a déclaré Frenkel. "Un outil polyvalent pour calculer les solubilités fait défaut depuis longtemps. La méthodologie sous-jacente était là, mais personne n'avait réellement créé un programme de travail."
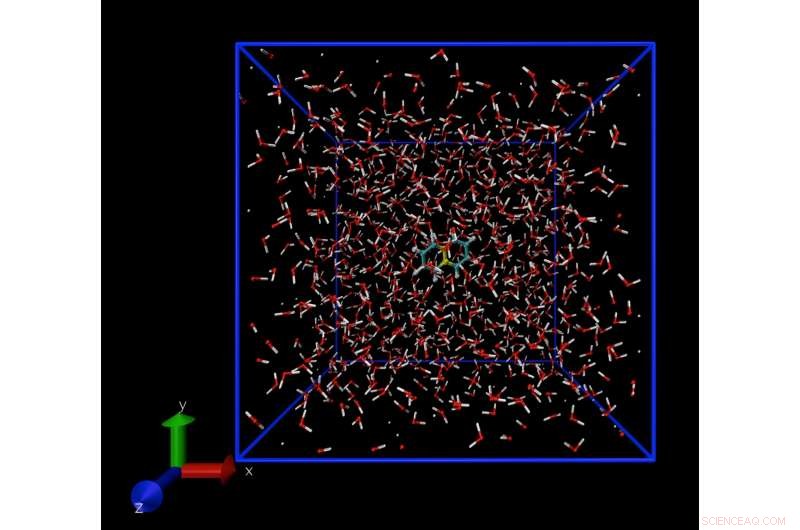
Un instantané d'une simulation de dynamique moléculaire montrant une seule molécule de naphtalène, dissous dans l'eau. La technique de simulation permet de calculer la concentration de molécules de naphtalène dans l'eau à la limite de solubilité. Crédit :Daan Frenkel, Université de Cambridge
Les logiciels développés par ce groupe utilisent des expressions thermodynamiques standards connues depuis le milieu du XIXe siècle, comme la pression de vapeur. L'approche exploite le fait que lorsqu'une phase solide ou liquide est en équilibre, leurs pressions de vapeur sont égales. Lorsqu'un liquide ou un solide sont chauffés, les molécules s'échappent et forment de la vapeur. Cette pression de vapeur peut être calculée à l'aide de modèles informatiques.
Par exemple, un morceau de sucre se dissolvant dans l'eau :les molécules de sucre existent soit à l'état solide (le morceau de sucre cristallin) soit complètement entourées de molécules d'eau une fois qu'elles se sont dissoutes. La quantité de sucre dans chacune des deux phases, solide et solution, est déterminé par l'énergie nécessaire pour déplacer les molécules de sucre entre ces phases. La solubilité peut être calculée en calculant la pression de vapeur des deux phases et en les égalant.
Pour modéliser la phase solide, les enquêteurs ont utilisé un modèle appelé cristal d'Einstein. Dans ce modèle, les molécules de soluté sans interaction sont placées sur un réseau et attachées à un point du réseau avec un ressort mathématique. La pression de vapeur du cristal est calculée en calculant le travail nécessaire pour désactiver les ressorts et activer les interactions entre les molécules captives.
Pour modéliser une molécule de soluté dissous, les enquêteurs ont utilisé un potentiel énergétique standard pour le solvant en question, qui était de l'eau dans les exemples utilisés pour tester leur logiciel, et calculé le travail en trois étapes. D'abord, une cavité dans le solvant est créée. Une molécule de soluté est ensuite insérée dans la cavité et, finalement, la cavité est réduite à la taille de la molécule de soluté. Cette procédure élimine un certain nombre d'erreurs et produit des estimations précises de la pression de vapeur et, Donc, la solubilité.
Dans le rapport de cette semaine, les chercheurs ont testé leur code sur le naphtalène dissous dans l'eau et ont prédit une solubilité qui se compare bien aux valeurs expérimentales. Les recherches futures se concentreront sur l'extension du logiciel afin qu'il puisse gérer des molécules de soluté plus grosses.