La conception de nouveaux composés ou alliages dont les surfaces peuvent être utilisées comme catalyseurs dans des réactions chimiques peut être un processus complexe qui repose largement sur l'intuition de chimistes expérimentés. Une équipe de chercheurs du MIT a conçu une nouvelle approche utilisant l'apprentissage automatique, qui supprime le besoin d'intuition et fournit des informations plus détaillées que ce que les méthodes conventionnelles peuvent pratiquement fournir.
Par exemple, en appliquant le nouveau système à un matériau déjà étudié depuis 30 ans par des moyens conventionnels, l'équipe a découvert que la surface du composé pourrait former deux nouvelles configurations atomiques qui n'avaient pas été identifiées auparavant, et qu'une autre configuration observée dans des travaux antérieurs est probablement instable.
Les résultats sont décrits dans la revue Nature Computational Science. , dans un article rédigé par Xiaochen Du, étudiant diplômé du MIT, les professeurs Rafael Gómez-Bombarelli et Bilge Yildiz, Lin Li, membre du personnel technique du MIT Lincoln Laboratory, et trois autres.
Les surfaces des matériaux interagissent souvent avec leur environnement d'une manière qui dépend de la configuration exacte des atomes à la surface, qui peut différer selon les parties de la structure atomique du matériau qui sont exposées. Pensez à un gâteau étagé contenant des raisins secs et des noix :selon la façon exacte dont vous coupez le gâteau, différentes quantités et dispositions des couches et des fruits seront exposées sur le bord de votre tranche.
L’environnement compte également. La surface du gâteau aura un aspect différent s'il est trempé dans du sirop, ce qui le rend humide et collant, ou s'il est mis au four, ce qui rend la surface croustillante et foncée. Cela s'apparente à la façon dont les surfaces des matériaux réagissent lorsqu'elles sont immergées dans un liquide ou exposées à des températures variables.
Les méthodes habituellement utilisées pour caractériser les surfaces des matériaux sont statiques et examinent une configuration particulière parmi des millions de possibilités. La nouvelle méthode permet d'estimer toutes les variations, sur la base de quelques calculs de principes premiers automatiquement choisis par un processus itératif d'apprentissage automatique, afin de trouver les matériaux présentant les propriétés souhaitées.
De plus, contrairement aux méthodes actuelles typiques, le nouveau système peut être étendu pour fournir des informations dynamiques sur la façon dont les propriétés de surface changent au fil du temps dans des conditions de fonctionnement, par exemple lorsqu'un catalyseur favorise activement une réaction chimique, ou pendant qu'une électrode de batterie est en charge ou déchargement.
La méthode des chercheurs, qu'ils appellent cadre de reconstruction automatique de surfaces, évite d'avoir à utiliser des exemples de surfaces triés sur le volet pour entraîner le réseau neuronal utilisé dans la simulation. Au lieu de cela, il commence par un seul exemple de surface coupée vierge, puis utilise un apprentissage actif combiné à un type d'algorithme de Monte-Carlo pour sélectionner les sites à échantillonner sur cette surface, en évaluant les résultats de chaque exemple de site pour guider la sélection du prochain. sites.
En utilisant moins de 5 000 calculs de principes fondamentaux, parmi des millions de compositions et configurations chimiques possibles, le système peut obtenir des prédictions précises des énergies de surface pour divers potentiels chimiques ou électriques, rapporte l'équipe.
"Nous étudions la thermodynamique", explique Du, "ce qui signifie que, sous différents types de conditions externes telles que la pression, la température et le potentiel chimique, qui peuvent être liés à la concentration d'un certain élément, [nous pouvons étudier] ce que est la structure la plus stable pour la surface ?"
En principe, déterminer les propriétés thermodynamiques de la surface d'un matériau nécessite de connaître les énergies de surface dans un arrangement atomique unique spécifique, puis de déterminer ces énergies des millions de fois pour englober toutes les variations possibles et capturer la dynamique des processus en cours. Bien qu'il soit possible en théorie de faire cela par ordinateur, "ce n'est tout simplement pas abordable" à l'échelle d'un laboratoire typique, dit Gómez-Bombarelli.
Les chercheurs ont pu obtenir de bons résultats en examinant seulement quelques cas spécifiques, mais cela ne suffit pas pour fournir une véritable image statistique des propriétés dynamiques impliquées, dit-il.
Grâce à leur méthode, Du déclare :« Nous disposons de nouvelles fonctionnalités qui nous permettent d'échantillonner la thermodynamique de différentes compositions et configurations. Nous montrons également que nous sommes capables d'y parvenir à moindre coût, avec moins d'évaluations coûteuses de l'énergie mécanique quantique. Et nous sont également capables de le faire pour des matériaux plus durs, notamment des matériaux à trois composants.
"Ce qui se fait traditionnellement dans ce domaine", dit-il, "c'est que les chercheurs, sur la base de leur intuition et de leurs connaissances, ne testent que quelques surfaces supposées. Mais nous effectuons un échantillonnage complet, et cela se fait automatiquement." Il dit que "nous avons transformé un processus qui était autrefois impossible ou extrêmement difficile en raison du besoin d'intuition humaine. Aujourd'hui, nous avons besoin d'une intervention humaine minimale. Nous fournissons simplement la surface immaculée, et notre outil s'occupe du reste."
Cet outil, ou ensemble d'algorithmes informatiques, appelé AutoSurfRecon, a été mis gratuitement à disposition par les chercheurs afin qu'il puisse être téléchargé et utilisé par n'importe quel chercheur dans le monde pour aider, par exemple, à développer de nouveaux matériaux pour catalyseurs, comme pour le production d'hydrogène « vert » comme carburant alternatif sans émissions, ou pour de nouveaux composants de batteries ou de piles à combustible.
Par exemple, explique Gómez-Bombarelli, lors du développement de catalyseurs pour la production d'hydrogène, « une partie du problème est que l'on ne comprend pas vraiment en quoi leur surface est différente de leur masse au cours du cycle catalytique. Il y a donc ce décalage entre l'apparence du matériau. comme quand il est utilisé et à quoi il ressemble lorsqu'il est préparé avant d'être mis en action."
Il ajoute qu'"en fin de compte, en catalyse, l'entité responsable de l'action du catalyseur est constituée de quelques atomes exposés à la surface, donc ce à quoi ressemble exactement la surface à l'heure actuelle est très important." P>
Une autre application potentielle consiste à étudier la dynamique des réactions chimiques utilisées pour éliminer le dioxyde de carbone de l’air ou des émissions des centrales électriques. Ces réactions fonctionnent souvent en utilisant un matériau qui agit comme une sorte d’éponge pour absorber l’oxygène, de sorte qu’il élimine les atomes d’oxygène des molécules de dioxyde de carbone, laissant derrière lui du monoxyde de carbone, qui peut être un carburant ou une matière première chimique utile. Le développement de tels matériaux "nécessite de comprendre ce que la surface fait avec les oxygènes et comment elle est structurée", explique Gómez-Bombarelli.
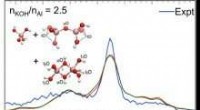
À l'aide de leur outil, les chercheurs ont étudié l'arrangement atomique de surface de l'oxyde de titane et de strontium, un matériau pérovskite, ou SrTiO3. , qui avait déjà été analysé par d’autres à l’aide de méthodes conventionnelles pendant plus de trois décennies, mais n’était toujours pas entièrement compris. Ils ont découvert deux nouveaux arrangements d'atomes à sa surface qui n'avaient pas été signalés auparavant, et ils prédisent qu'il est en fait peu probable qu'un arrangement signalé se produise.
"Cela montre que la méthode fonctionne sans intuitions", explique Gómez-Bombarelli. "Et c'est bien parce que parfois l'intuition est fausse, et ce que les gens pensaient être le cas s'avère ne pas l'être." Ce nouvel outil, a-t-il déclaré, permettra aux chercheurs d'être plus exploratoires et d'essayer un plus large éventail de possibilités.
Maintenant que leur code a été rendu public par la communauté dans son ensemble, dit-il, "nous espérons qu'il servira d'inspiration pour des améliorations très rapides" par d'autres utilisateurs.
L'équipe comprenait James Damewood, titulaire d'un doctorat. étudiante au MIT, Jaclyn Lunger Ph.D., qui travaille maintenant à Flagship Pioneering, et Reisel Millan, un ancien postdoctorant qui travaille maintenant à l'Institut de technologie chimique en Espagne.
Plus d'informations : Simulations accélérées par apprentissage automatique pour permettre une reconstruction de surface sans heuristique, Nature Computational Science (2023). DOI :10.1038/s43588-023-00571-7
Informations sur le journal : Science informatique de la nature
Fourni par le Massachusetts Institute of Technology
Cette histoire est republiée avec l'aimable autorisation de MIT News (web.mit.edu/newsoffice/), un site populaire qui couvre l'actualité de la recherche, de l'innovation et de l'enseignement du MIT.