Crédit :Université de Californie, San Francisco
La découverte de médicaments peut faire penser à des images de blouses blanches et de pipettes, mais quand Henry Lin, Doctorat, a récemment entrepris de trouver un meilleur opioïde avec moins d'effets secondaires, sa première étape consistait à allumer les ordinateurs.
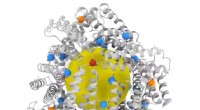
En utilisant un programme appelé DOCK, il a téléchargé une structure cristalline du récepteur opioïde trouvé dans le cerveau et a accédé à une bibliothèque virtuelle de 3 millions de composés qui pourraient se lier à une "poche" chimique sur le récepteur. La plupart des médicaments - des antibiotiques aux antidépresseurs - agissent en se liant à des sites spécifiques sur les protéines, mais pour être efficace, ils doivent s'adapter parfaitement.
Le programme a fait tourner chaque composé, considéré la souplesse de ses différents appendices, et après avoir testé en moyenne 1,3 million de configurations par composé, les a classées en fonction de leur potentiel de liaison. Le processus, fonctionnant sur des ordinateurs connectés à de puissants processeurs, a pris environ deux semaines.
Étudiant diplômé à l'époque, Lin a travaillé avec son conseiller Brian Shoichet, Doctorat, professeur de chimie pharmaceutique à l'UC San Francisco School of Pharmacy, et Aashish Manglik, Doctorat, de l'Université de Stanford pour se classer parmi les 2 premiers, 500 composés pour des facteurs supplémentaires et 23 sélectionnés pour des tests expérimentaux dans des cellules vivantes - sarraus de laboratoire et pipettes.
De plus en plus, les chercheurs se tournent vers les expériences virtuelles pour les premières étapes du développement de médicaments. Avec des ordinateurs toujours plus rapides, la phase précoce et en grande partie d'essais et d'erreurs du développement d'un médicament peut être réduite à quelques jours, et avec des bibliothèques en ligne de composés en constante expansion, les criblages de médicaments peuvent englober, au sens propre, toute la chimie connue dans le monde.
Forces et limites
Les chercheurs sont prudents quant au potentiel de la découverte de médicaments informatiques - seule une petite fraction des composés prometteurs fonctionnent réellement lorsqu'ils sont testés dans la vie réelle - mais ils disent que l'une de ses forces est de révéler des composés entièrement nouveaux en tant que candidats-médicaments.
Shoichet se spécialise dans une méthode de calcul populaire connue sous le nom d'amarrage moléculaire. "L'amarrage s'intègre dans les premières recherches de découverte, à trouver de nouveaux départs, " il a dit.
La recherche de son équipe pour le nouvel opioïde illustre à la fois les forces et les limites de la découverte informatique de médicaments.
En réalité, les candidats opioïdes initiaux identifiés grâce à l'amarrage moléculaire n'ont donné que des résultats modestes lors des tests expérimentaux. "Toujours, l'activité qu'ils avaient était hautement reproductible et les molécules étaient très nouvelles, présage d'une nouvelle biologie, " dit Shoichet.
L'équipe a amarré une autre série de composés avec des structures similaires et a testé les meilleurs buteurs. Avec des collaborateurs de l'Université de Caroline du Nord, Chapel Hill et l'Université Friedrich Alexander en Allemagne, ils ont identifié le composé le plus puissant et optimisé sa pharmacologie avec une élaboration synthétique guidée par ordinateur.
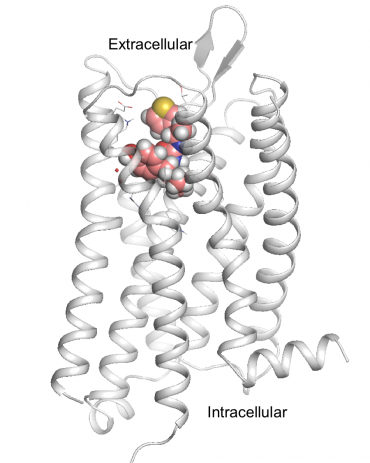
PZM21, le nouveau, candidat médicament opioïde plus sûr, est montré amarré sur le récepteur de la morphine du cerveau, le récepteur mu-opioïde. Crédit :Anat Levit
Ce composé gagnant, nommé PZM21, est chimiquement différent de tout ce qui est actuellement utilisé et n'a peut-être pas été trouvé par des méthodes plus traditionnelles. C'est un composé entièrement conçu par ordinateur qui est plus puissant que la morphine. Chez la souris, il bloquait efficacement la douleur sans les effets secondaires habituels de la suppression respiratoire et de la constipation et semblait même être moins addictif.
L'amarrage n'est pas une solution miracle, mais c'est devenu un puissant point de départ pour longtemps, processus interdisciplinaire de développement de médicaments. Parmi ses contributions majeures figurent les inhibiteurs de protéase qui ont contribué à faire du VIH une maladie traitable. Les chercheurs utilisent également l'amarrage pour cribler des candidats médicaments pour le traitement du cancer du sein, hépatite C, hypertension, Staphylocoque, le virus du SRAS et la grippe.
Technologie pionnière à l'UCSF
L'amarrage moléculaire a été lancé il y a trois décennies par un jeune chimiste physique de l'UCSF nommé Tack Kuntz, Doctorat, maintenant professeur émérite à l'École de pharmacie. Lorsque Kuntz est arrivé sur le campus au début des années 1970, l'approche traditionnelle de la découverte de médicaments prévalait encore.
Comme Kuntz l'a décrit, le processus reposait sur le hasard et très peu de théorie :« Vous sortez et trouvez de nouveaux composés naturels et les rapportez pour les tester dans un laboratoire. Il suffit de mettre des produits chimiques ensemble avec un organisme et de voir ce qui se passe. »
Les chimistes pharmaceutiques ont à peine pensé aux détails moléculaires de la façon dont les médicaments interagissaient avec le corps. De nombreux médicaments, dont les premiers antibiotiques, avait été découvert par hasard, mais Kuntz, ayant vu la nouvelle compréhension moléculaire balayer le domaine de la biologie, a estimé qu'il était temps pour une mise à jour similaire en pharmacologie.
"La vision de la biologie basée sur les cibles - que vous pouvez comprendre la biologie à travers des protéines et des produits génétiques indépendants - avait déjà pris le dessus, mais la pharmacologie avait une décennie de retard, " dit Shoichet, qui était un étudiant diplômé dans le laboratoire de Kuntz dans les années 1980.
Kuntz et ses collègues ont commencé à travailler vers une approche plus rationnelle de la conception de médicaments dans laquelle ils ont essayé d'identifier des composés qui pourraient s'adapter à des récepteurs spécifiques sur des protéines, comme trouver la pièce manquante d'un puzzle. En 1982, ils ont publié un article décrivant le premier programme d'amarrage moléculaire qui pourrait « explorer des alignements géométriquement réalisables de ligands et de récepteurs de structure connue ».
Kuntz en a envoyé 10, 000 exemplaires de ce premier programme d'amarrage aux chercheurs de tout le pays. Bientôt, d'autres chercheurs développaient des programmes informatiques similaires et l'enthousiasme s'est rapidement propagé en dehors du milieu universitaire. Dans les années 1990, chaque grande entreprise pharmaceutique avait ouvert une unité de découverte informatique de médicaments.
Rattraper une idée
Malgré l'enthousiasme initial, cependant, la découverte de médicaments informatiques n'a pas conduit à des résultats rapides. L'idée de Kuntz était arrivée en avance sur son temps. Il faudrait des décennies d'avancées progressives en biologie moléculaire, technologie de l'imagerie et de l'informatique, avant que la découverte de médicaments informatiques ne commence à tenir ses promesses.

Tack Kuntz, Doctorat, et ses collègues ont publié en 1982 un article décrivant le premier programme d'amarrage moléculaire qui pourrait « explorer des alignements géométriquement réalisables de ligands et de récepteurs de structure connue ». Crédit :Université de Californie, San Francisco
Une limitation majeure dans les années 1990 était le manque de structures protéiques connues. Sans ceux-ci, il y avait peu de cibles pour lesquelles trouver des drogues. Dans les décennies qui ont suivi, des milliers de structures protéiques de cibles médicamenteuses potentielles ont été révélées par cristallographie aux rayons X et imagerie par résonance magnétique nucléaire.
La découverte du nouveau candidat opioïde, par exemple, n'a été possible qu'en raison des structures cristallines récemment déterminées des récepteurs couplés aux protéines G, une famille de protéines qui comprend le récepteur opioïde.
Les bibliothèques virtuelles de composés ont également connu une croissance exponentielle. En 1991, une base de données peut en contenir 55, 000 composés ; maintenant ils en contiennent des dizaines de millions. "La portée de la chimie que nous échantillonnons a augmenté à peu près au même rythme que la loi de Moore, " a déclaré Shoichet. "Il y a une faim insatiable pour de plus en plus de molécules."
Les programmes d'accueil d'aujourd'hui sont capables de modéliser de manière réaliste les interactions au niveau atomique entre un médicament et sa cible, mais certains détails délicats - tels que la façon dont les forces atomiques changent lorsqu'une molécule de médicament déplace l'eau au site de liaison - restent des défis permanents sur le terrain.
Promesses et preuves
L'amarrage moléculaire n'est pas la seule forme de conception de médicaments informatisée. À l'UCSF Institute for Computational Health Sciences (ICHS), des dizaines de chercheurs explorent une myriade de méthodes informatiques pour faire avancer la recherche médicale.
Michael Keiser, Doctorat, membre de l'ICHS et professeur assistant à l'Institut des maladies neurodégénératives, étudie des médicaments qui atteignent plusieurs cibles moléculaires à la fois, comme si vous frappiez un accord plutôt qu'une seule note. Cette action multi-cibles a longtemps été considérée comme la cause d'effets secondaires involontaires, mais peut également être dirigé pour traiter des maladies complexes.
Ce n'est qu'au début des années 2000 que les chercheurs en sont venus à reconnaître que de nombreux médicaments existants agissent sur plus d'une cible - les antipsychotiques, par exemple, qui frappent à la fois les récepteurs de la sérotonine et de la dopamine. Ils conçoivent maintenant intentionnellement des médicaments pour le faire.
"Pour certaines maladies qui n'ont pas encore de traitement, c'est peut-être parce qu'il n'y a pas une seule protéine que vous devez activer ou désactiver ; Et si le médicament devait toucher plusieurs cibles à la place ?", a déclaré Keiser, qui était un étudiant diplômé de Shoichet.
Dans son laboratoire, Keizer utilise des méthodes informatiques pour identifier les modèles chimiques parmi les médicaments qui se lient au même ensemble de cibles et trouver de nouveaux composés qui ont une pharmacologie correspondante. Cette approche informatique peut reconnaître des similitudes entre les composés que des analyses plus conventionnelles manqueraient. Keizer se tourne désormais vers la technologie de l'intelligence artificielle, connu sous le nom d'apprentissage en profondeur, pour une meilleure reconnaissance des formes.
Alors même que les méthodes de calcul décollent, leur preuve est toujours dans le monde réel - dans les cellules, modèles animaux, et finalement en clinique. "Pendant un certain temps, il était courant de publier des articles contenant des prédictions sur les activités d'une petite molécule, mais pas de test réel de ces prédictions, parce que les expériences pour le faire étaient coûteuses, difficile ou ésotérique, " dit Keiser.
Le besoin de collaboration étant devenu évident, le partenariat entre la prédiction informatique et les expériences en laboratoire humide s'est sensiblement renforcé au cours de la dernière décennie, dit Keiser. "Après tout, comment pouvez-vous améliorer vos prédictions si vous n'êtes pas sûr de celles qui sont fausses ? »