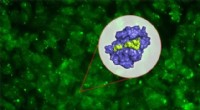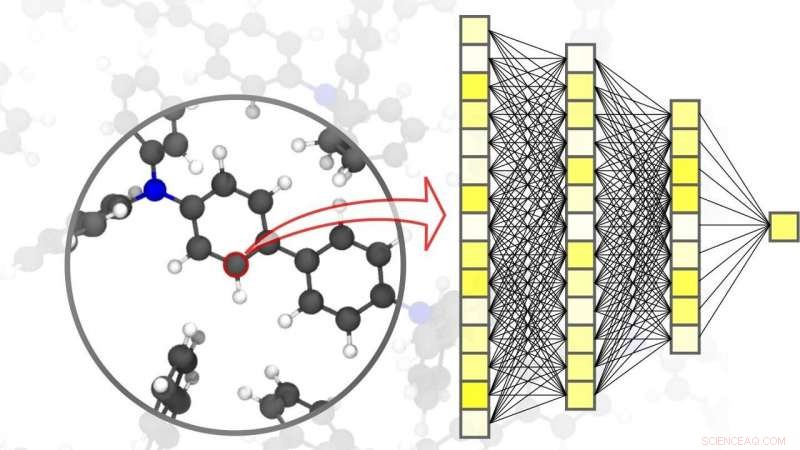
Les réseaux de neurones permettent des simulations précises en science des matériaux, jusqu'au niveau des atomes individuels. Crédit :Pascal Friederich, TROUSSE
Recherche, développement, et la production de nouveaux matériaux dépendent fortement de la disponibilité de méthodes de simulation rapides et précises à la fois. Apprentissage automatique, dans lequel l'intelligence artificielle (IA) acquiert et applique de manière autonome de nouvelles connaissances, permettra bientôt aux chercheurs de développer des systèmes matériels complexes dans un environnement purement virtuel. Comment cela marche-t-il, et quelles applications en bénéficieront ? Dans un article publié dans le Matériaux naturels journal, un chercheur du Karlsruhe Institute of Technology (KIT) et ses collègues de Göttingen et de Toronto expliquent tout.
La numérisation et la virtualisation deviennent de plus en plus importantes dans un large éventail de disciplines scientifiques. L'une de ces disciplines est la science des matériaux :la recherche, développement, et la production de nouveaux matériaux dépendent fortement de la disponibilité de méthodes de simulation rapides et précises à la fois. Cette, à son tour, est bénéfique pour un large éventail d'applications différentes - des systèmes de stockage d'énergie efficaces, telles que celles indispensables à l'utilisation des énergies renouvelables, aux nouveaux médicaments, pour le développement desquels une compréhension des processus biologiques complexes est requise. L'IA et les méthodes d'apprentissage automatique peuvent faire passer les simulations en sciences des matériaux au niveau supérieur. « Par rapport aux méthodes de simulation conventionnelles basées sur des calculs de mécanique classique ou quantique, l'utilisation de réseaux de neurones spécifiquement adaptés aux simulations de matériaux nous permet d'obtenir un avantage de vitesse significatif, " explique le physicien et expert en IA le professeur Pascal Friederich, Chef du groupe de recherche AiMat—Intelligence artificielle pour les sciences des matériaux à l'Institut d'informatique théorique (ITI) du KIT. "Avec des systèmes de simulation plus rapides, les scientifiques pourront développer des systèmes matériels plus grands et plus complexes dans un environnement purement virtuel, et de les comprendre et de les optimiser jusqu'au niveau atomique."
Haute précision de l'atome à la matière
Dans un article publié dans Matériaux naturels , Pascal Friederich, qui est également chef de groupe associé de la division Nanomatériaux par conception guidée par l'information à l'Institut de nanotechnologie du KIT (INT), présente, avec des chercheurs de l'Université de Göttingen et de l'Université de Toronto, un aperçu des principes de base de l'apprentissage automatique utilisés pour les simulations en sciences des matériaux. Cela inclut également le processus d'acquisition de données et les méthodes d'apprentissage actif. Les algorithmes d'apprentissage automatique permettent non seulement à l'intelligence artificielle de traiter les données d'entrée, mais aussi pour trouver des modèles et des corrélations dans de grands ensembles de données, apprendre d'eux, et faire des prédictions et des décisions autonomes. Pour les simulations en science des matériaux, il est important d'obtenir une grande précision sur différentes échelles de temps et de taille, allant de l'atome à la matière, tout en limitant les coûts de calcul. Dans leur article, les scientifiques discutent également de diverses applications actuelles, telles que les petites molécules organiques et les grandes biomolécules, solide structurellement désordonné, liquide, et matières gazeuses, ainsi que des systèmes cristallins complexes - par exemple, charpentes organométalliques utilisables pour le stockage de gaz ou pour la séparation, pour capteurs ou pour catalyseurs.
Encore plus de vitesse avec les méthodes hybrides
Pour étendre encore les possibilités de simulations de matériaux à l'avenir, les chercheurs de Karlsruhe, Göttingen, et Toronto suggèrent le développement de méthodes hybrides :celles-ci combinent des méthodes d'apprentissage automatique (ML) et de mécanique moléculaire (MM). Les simulations MM utilisent des champs de force afin de calculer les forces agissant sur chaque particule individuelle et ainsi prédire les mouvements. Comme les potentiels des méthodes ML et MM sont assez similaires, une intégration étroite avec des zones de transition variables est possible. Ces méthodes hybrides pourraient accélérer considérablement la simulation de grandes biomolécules ou de réactions enzymatiques à l'avenir, par exemple.