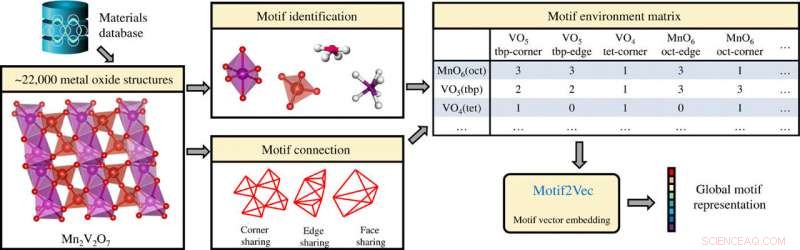
Extraction d'informations sur les motifs de structure dans des composés cristallins inorganiques (oxydes métalliques) et génération de représentations globales de motifs à l'aide de la matrice d'environnement de motifs. Crédit: Avancées scientifiques , doi:10.1126/sciadv.abf1754
Les principes physiques peuvent être incorporés dans une architecture d'apprentissage automatique en tant que configuration fondamentale pour développer l'intelligence artificielle pour les matériaux inorganiques. Dans un nouveau rapport maintenant sur Avancées scientifiques , Huta R. Banjade, et une équipe de recherche en physique, L'informatique et les sciences de l'information et les nanosciences aux États-Unis et en Belgique ont proposé des motifs de structure dans des cristaux inorganiques pour servir d'entrée centrale à un cadre d'apprentissage automatique. L'équipe a démontré comment la présence de motifs de structure et leurs connexions dans un grand ensemble de composés cristallins pouvaient être converties en représentations vectorielles uniques via un algorithme d'apprentissage non supervisé. Ils y sont parvenus en créant un cadre d'apprentissage centré sur les motifs en combinant des informations sur les motifs avec des réseaux de neurones à base d'atomes pour former un réseau à double graphe atome-motif (AMDNet). La configuration a prédit avec précision la structure électronique des oxydes métalliques tels que les bandes interdites. Le travail illustre une méthode pour concevoir des architectures d'apprentissage de réseaux de neurones graphiques pour étudier des matériaux complexes au-delà des propriétés physiques des atomes.
Méthodes de ML
Les méthodes d'apprentissage automatique (ML) peuvent être combinées à des données massives sur les matériaux pour accélérer la découverte et la conception rationnelle de composés fonctionnels à l'état solide. L'apprentissage supervisé peut conduire à des prédictions de propriétés matérielles, y compris la stabilité de phase et la nature cristalline, efficace pour les simulations de dynamique moléculaire. Les motifs de structure peuvent être créés conformément à la première règle de Pauling, en formant un polyèdre coordonné d'anions autour de chaque cation dans un composé pour se comporter comme des blocs de construction fondamentaux qui sont fortement corrélés avec les propriétés des matériaux. Par exemple, les motifs de structure dans les composés cristallins peuvent jouer un rôle essentiel pour déterminer les propriétés des matériaux dans diverses applications techniques et scientifiques. Dans ce travail, Banjade et al. incorporé des informations de motif de structure dans un cadre d'apprentissage automatique (ML). Les scientifiques ont combiné les informations sur les motifs avec des réseaux de neurones convolutifs de graphes pour développer une architecture d'apprentissage en profondeur centrée sur les motifs connue sous le nom de réseau à double graphe atome-motif (AMDNet). La précision de la structure a dépassé celle d'un réseau de graphes à base d'atomes de pointe existant pour prédire les structures électroniques des matériaux cristallins inorganiques.
La projection stochastique voisine de distribution t des vecteurs de motifs construits en utilisant la matrice d'environnement de motifs. Les groupes de motifs 1 à 4 sont associés à différents types de motifs dont (1) cube, (2) cuboctaèdre, (3) octaèdre, et (4) un mélange de tétraèdre (en magenta) et de plan carré (en reste). t-SNE, plongement voisin stochastique à distribution t. Crédit: Avancées scientifiques , doi:10.1126/sciadv.abf1754 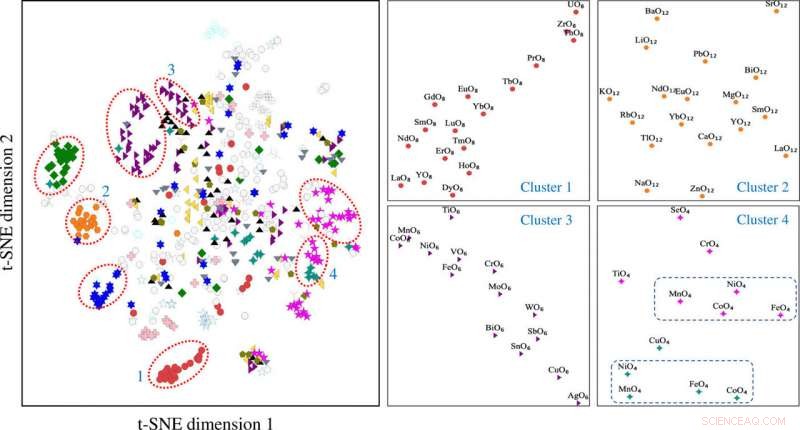
Un algorithme d'apprentissage non supervisé Atom2Vec peut comprendre les représentations vectorielles de grande dimension des atomes en codant les propriétés de base des atomes sur la base d'une vaste base de données de formules chimiques. Banjade et al. axé sur les oxydes métalliques binaires et ternaires qui constituent un espace matériel vaste et diversifié où les structures cristallines sont caractérisées par la coordination cation-oxygène. Pour extraire les informations de motif de structure, l'équipe a utilisé la méthode d'identification de l'environnement local développée par Waroquiers et al. tel qu'implémenté par le code Pymatgen. L'équipe a identifié trois types différents de connectivité entre un motif et son motif voisin; y compris le partage interne (un atome partagé), partage de bord (deux atomes partagés), et partage de visage (trois atomes ou plus partagés). Les scientifiques ont ensuite proposé un algorithme d'apprentissage pour tirer parti du processus de collecte de données de motif et ont efficacement converti chaque ligne de la matrice d'environnement de motif en un vecteur de grande dimension pour représenter un motif de structure unique. Ils ont ensuite extrait des informations sur les motifs pour le processus d'apprentissage à l'aide d'un réseau convolutif de graphes. L'équipe visait à identifier les modèles et les informations de regroupement pour ces vecteurs de motifs de grande dimension afin d'influencer les propriétés matérielles complexes des composés d'oxyde. Ils ont visualisé les données de grande dimension à l'aide du plongement voisin stochastique à distribution t (t-SNE) - une technique de réduction de dimensionnalité non linéaire.
Utilisation des informations de motif dans les réseaux de neurones graphiques.
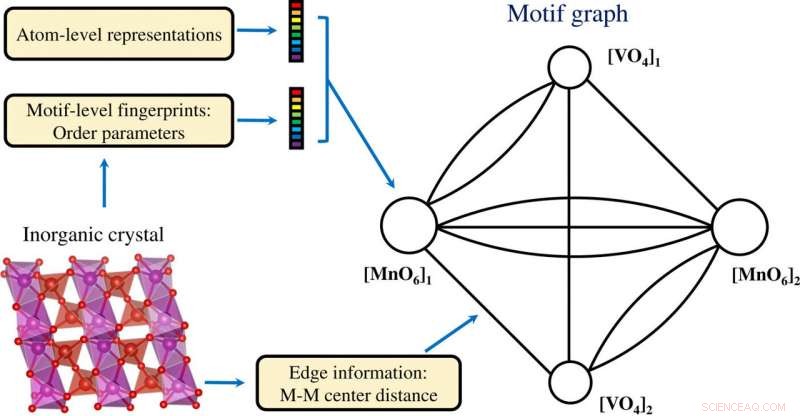
Construction d'un graphe de motifs basé sur des informations au niveau de l'atome et au niveau du motif codées dans un cristal inorganique. Crédit: Avancées scientifiques , doi:10.1126/sciadv.abf1754
Les scientifiques ont obtenu des données vectorielles de motifs projetées en deux dimensions à l'aide du processus t-SNE. Ils ont noté des groupes distincts en fonction des types de motifs. Les propriétés chimiques des éléments formant les motifs ont joué un rôle clé lors de la formation des amas. Par exemple, Les motifs à base de lanthanide ont formé différents groupes sur la base du type de motif et les motifs à base d'yttrium sont restés proches des motifs à base de lanthanide en raison de leurs similitudes chimiques. Les motifs associés au zinc et au magnésium se sont également regroupés. Les résultats basés sur l'apprentissage non supervisé ont soutenu les motifs de structure pour servir d'entrées essentielles pour les composés cristallins porteurs d'informations élémentaires et structurelles. L'équipe a ensuite utilisé les informations de motif de structure comme entrée essentielle à un réseau de neurones graphiques (GNN) pour prédire les propriétés physiques des matériaux. La plupart des réseaux de graphes appliqués aux matériaux cristallins. Pour permettre une architecture d'apprentissage des représentations graphiques au niveau de l'atome et au niveau du motif des matériaux, Banjade et al. ont proposé qu'AMDNet puisse être construit pour améliorer le processus d'apprentissage et améliorer la précision de la prédiction des propriétés de la structure électronique des oxydes métalliques. Dans les graphiques de motifs, les chercheurs ont codé les informations au niveau de l'atome et du motif dans chaque nœud et ont construit le graphe de motifs, y compris la connectivité étendue, angle, paramètres de distance et d'ordre à l'aide de la robocristallographie du package Python.
AMDNet
Dans l'architecture AMDNet proposée, Banjade et al. incorporé des informations de motif dans un cadre d'apprentissage de réseau de graphes pour générer des graphes de motifs et des graphes d'atomes représentant des composés avec différentes cardinalités d'arêtes et de nœuds pour combiner les informations avant de faire des prédictions. Pour chaque matériau, l'équipe a généré un graphe d'atomes et un graphe de motifs. Ils ont ensuite utilisé 22, 606 oxydes métalliques binaires et ternaires de la base de données Materials Project pour tester l'efficacité du modèle proposé et se sont concentrés sur la prédiction des bandes interdites, un problème de structure électronique complexe. Les résultats ont montré la supériorité d'AMDNet lors de la prédiction de la bande interdite par rapport aux réseaux précédents. Le modèle a également montré des performances supérieures lors d'une tâche de classification des métaux par rapport aux non-métaux. Le travail a montré les efforts initiaux pour incorporer des informations matérielles de haut niveau dans les modèles d'apprentissage en profondeur pour les matériaux à l'état solide.
Prédictions de l'architecture et des propriétés des matériaux AMDNet. (A) Démonstration de l'architecture d'apprentissage du réseau à double graphe atome-motif proposé (AMDNet) pour l'apprentissage efficace des structures électroniques et d'autres propriétés matérielles des matériaux cristallins inorganiques. (B) Comparaison des bandes interdites prévues et réelles [à partir des calculs de la théorie fonctionnelle de la densité (DFT)] et (C) comparaison des énergies de formation prévues et réelles (à partir des calculs DFT) dans l'ensemble de données de test avec 4515 composés. Crédit: Avancées scientifiques , doi:10.1126/sciadv.abf1754 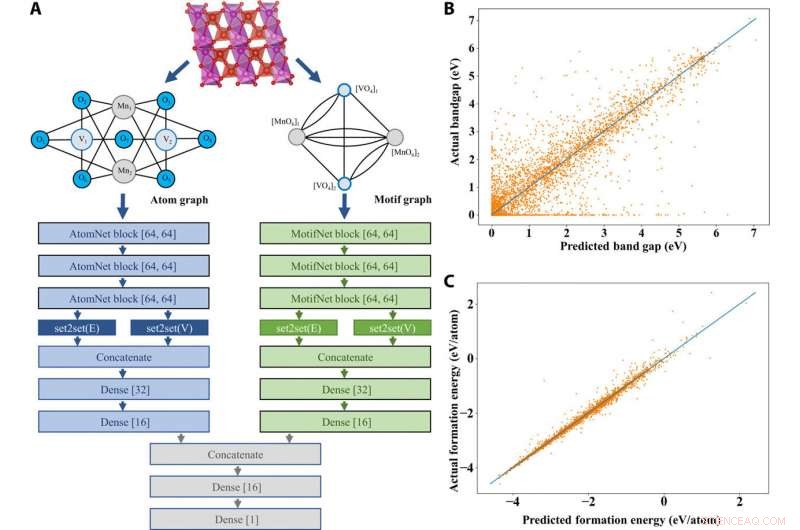
De cette façon, Huta R. Banjade et ses collègues ont montré comment les motifs de structure dans les structures cristallines pouvaient être combinés avec des méthodes d'apprentissage automatique non supervisées et supervisées pour améliorer la représentation efficace des systèmes de matériaux à l'état solide. Pour les structures électroniques complexes, l'équipe a inclus les informations de connexion de structure et de motif dans un modèle AMDNet pour surpasser les réseaux existants et prédire les bandes interdites électroniques et les tâches de classification des métaux par rapport aux non-métaux. Ce cadre d'apprentissage général peut être utilisé pour prédire les propriétés d'autres matériaux, y compris les propriétés d'état mécanique et excité à travers des matériaux bidimensionnels et des cadres métal-organiques.
© 2021 Réseau Science X














